基于密度泛函理论下H2S 在单原子催化剂V/Ti2CO2 上的分解机理研究
2021-09-01周君慧敖志敏安太成
周君慧,敖志敏,安太成
广东工业大学环境健康与污染控制研究院,环境科学与工程学院,广东省环境催化与健康风险控制重点实验室和广州市环境催化与污染控制重点实验室,广州 510006
1 引言
随着工业化的加速推进,大量的化石燃料被广泛使用并伴随着大量的有毒气体排放,造成大气环境污染现象日益加剧,严重制约着经济的可持续发展1。其中硫化氢(H2S)气体作为化学工业排放的最具毒性的恶臭化合物之一,其主要来源是天然气开采、高温焦化以及原油炼制等工业过程2,3。据报道罗马尼亚黑海油气公司已经于2018年开始黑海(欧亚内陆海)天然气的生产,其底部的含H2S的酸性天然气储藏量大约为4.6 × 109吨4。硫化氢的排放不仅对生态环境会造成破坏,而且对人类也会产生较大威胁,在人体暴露在硫化氢浓度高于41.8 mg·m−3时,会损伤人体嗅觉,与此同时,会导致细胞缺氧,硫化氢会经呼吸迅速进入血液中,造成细胞缺氧1,当浓度超过975.7 mg·m−3时甚至会导致机体死亡4。鉴于此,含硫气体必须经过处理,达到相应的工业排放标准之后才能排放到大气中5,6。因此,使用合适的方法去除硫化氢成为工程人员亟待解决的问题。
据报道,石化行业的原燃料气体硫化氢的浓度在1393.9-1951.4 mg·m−3范围内,如此高浓度的硫化氢不仅会对人体造成危害,而且可能会腐蚀工艺中的管道和设备。其中,去除H2S主要是为了保护燃气轮机免受碱硫酸盐形成的高温腐蚀,因此部分工业将H2S浓度限制在27.9 mg·m−3以内。迄今为止,有关H2S的去除已经有大量的研究报道,其中使用非贵金属催化剂有β-CuxV2O55、SnO2/QDs-C607、CuO/SnO28以及WO39等,虽然这些廉价的催化剂从实用的角度看很有吸引力,但是这些催化剂的操作温度都在400 K以上,处理效果不是很显著。使用贵金属催化剂的有Pt/graphene(反应能垒为0.37 eV)10和Pd(100) (反应能垒为0.28 eV)11,虽然这些催化剂处理H2S的效率较高,但是贵金属的使用限制了它们的广泛应用。这些催化剂处理硫化氢涉及到的方法有吸收法12、吸附法13以及克劳斯法1,5,14。其中,吸收和吸附法只是简单的将H2S从气相分别转化至液相和固相,并没有从本质上将污染物分解。而克劳斯法中是工业上处理硫化氢气体的一种较为广泛使用的方法,该方法不仅能够将该H2S去除,而且能从含硫气体中回收单质硫。该方法主要涉及到以下反应:
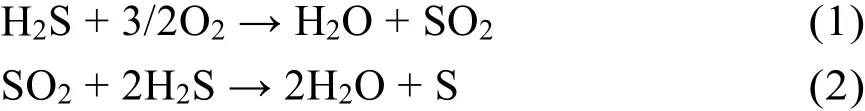
然而,这个方法的限制之处在于这个反应过程(1)需要大量的热(反应温度在1200-1500 K1,而且反应(2)中单质硫在氧气过量的条件下会生成对环境造成二次污染的气体SO2。因此,H2S气体去除经常遇到的挑战是副产物SO2的干扰,这对于提高克劳斯工艺的处理效率和H2S气体处理的成本不仅起着至关重要的作用,而且这也是工业大规模处理硫化氢迫切需要解决的问题。为了应对这些技术挑战,需要开发稳定、低成本、具有较高反应活性并且可支持大规模生产的催化剂15-19。此外,上述的大多数技术都处于研究的早期阶段,离商业化还很遥远,仍然需要完善基础应用研究来提高技术成熟度。
近年来,单原子催化剂(single-atom catalysts,SACs)以其高活性、高效率以及高稳定性等特点引起了人们的广泛关注。对于硫化氢的去除在实验和理论上已经有一些研究,其中不仅N/TiO220以及Ni21,22、Fe23和Pt10等单原子负载于二维材料上的单原子催化剂对H2S都有较高的去除效果,一些贵金属(Pd24,25、Au26和Ag27)的表现也是可圈可点。然而贵金属催化剂因其较高的成本限制了它们的发展,尤其是工业上的大规模应用。SACs的稳定性不仅和负载的单原子有关系,载体的选择也是不可或缺。以往的研究表明二维(2D)材料通常具有独特的物理和化学性质,尤其是具有较高的比表面积28,这对于催化反应至关重要。不仅如此,金属原子负载在二维材料上,能够以单原子形式稳定分散在二维材料表面,而不会产生团簇现象,并为催化反应提供一个高活性位点28,29。近些年来,基于二维材料作为载体的SACs在一氧化碳催化氧化29-32和甲醛降解28,33,34等环境领域都有较大的应用。因此,开发低成本的致力于降解硫化氢的二维SACs是我们这个工作的主要研究内容。
在众多2D材料中,MXene作为一种很有发展前途的二维材料,在能源35-37和催化领域28-30,38,39等方面得到了广泛的研究,而且结果表明MXene在催化反应中具有较好的稳定性、活性和选择性29,39-44。MXene是由MAX相经氢氟酸(HF)蚀刻而得到,其中M指的是前过渡金属,A则代表第三和第四主族的元素,X是专指碳或者氮元素37,45。但是本征MXene总会不可避免地被HF溶液中的F或者OH基团以及空气中的O所功能化,从而形成Mn+1CnT2(T = F、OH或者O)37,46。Li等47报道了表面被O所功能化的MXene比被OH/F基团所功能化的结构更稳定。此外,Zhang等报道说在高温处理时,表面被OH所功能化的MXene会转变为O钝化的MXene材料29。因此,被O所功能化的MXene更容易被制备出来。而Ti2CO2作为一种已经被制备出来的典型MXene,不仅拥有较高的比表面积和较强的化学稳定性,而且表现出较好的表面性能,尤其表现在其和金属之间具有较强的相互作用29,48,并且在催化领域有很好的表现。近年来,Ti2CO2广泛应用于CO2催化还原39、CO催化氧化29、N2还原49、HER和OER50,51等领域中。因此,我们提出将Ti2CO2作为SACs的载体,将单原子负载在其表面并研究其催化活性,我们期待该催化剂在硫化氢分解应用中有所建树。
在本工作中,我们将使用密度泛函理论(Density Functional Theory,DFT)计算H2S在单原子催化剂(单原子负载的Ti2CO2)表面上的处理过程及其反应机理进行研究。通过DFT计算,研究相应的吸附行为、电子转移过程及机理,材料或者化学物质的电子构型,并阐述化学反应的热力学和动力学过程。相关研究表明:单原子的电负性52越低(Ti、V和Cr),与载体材料的结合能越强28,而且这三种金属都是第四周期上相邻的过渡金属,具有类似的电子结构。因此我们选择Ti、V和Cr三种过渡金属负载于Ti2CO2表面上作为SACs来探索H2S的解离机理,我们将对催化剂与H2S之间的电荷转移、分波态密度(PDOS)以及H2S的分解反应路径进行分析。本研究将为开发高效去除硫化氢的催化剂提供理论指导,并试图阐明MXene在污染物催化降解领域的应用前景。
2 计算方法
本研究采用基于密度泛函理论的Materials Studio软件中的Dmol3模块53进行计算。交换相关泛函采用广义梯度近似法GGA-PBE54。计算中采用了基于Grimme的DFT-D校正方法考虑范德华力的影响55。基组采用双数值极化函数(DNP)56,内核电子处理方法采用引入相对论校正的半经验赝势(DSPPs)57。自洽迭代收敛偏差为10−5Ha (1 Ha =27.21 eV),原子间相互作用力和最大位移分别是0.02 Ha·nm−1和5 × 10−4nm,最大迭代次数为100次。为了加速自洽迭代的收敛速度,采用热拖尾效应方法应用到占据轨道上,拖尾值为0.005 Ha。所有计算都在倒格子空间中执行。
我们采用Dmol3模块中的线性同步变换/二次同步变换(Complete LST/QST)58和微扰弹性带(NEB)59方法来研究硫化氢分子在催化剂表面的解离的最小能量路径。在计算时,我们模拟构建的三维周期性超晶胞包含一个3 × 3 × 1的Ti2CO2单元晶胞,与此同时,我们在Ti2CO2上表面的氧原子层上方增加了1.5 nm的真空层以此消除周期性层间的相互作用39。为了得到更精确的计算结果,所有的原子都允许弛豫,直到原子间相互作用力小于0.02 Ha·nm−1。在对超胞几何构型进行优化计算时,k点取样密度为2 × 2 × 1 (单胞为5 × 5 × 1),在进行态密度(DOS)和能带(Band gap)计算时则采用密度更高的3 × 3 × 1的k点网格。所有的计算精度等级全部采用Fine,以保证计算结果的准确性。分子的化学反应活性和电荷分布有关,因此,我们采用布居分析(Population analysis)来计算体系中所有分子或者原子的Hirshfeld电荷分布情况60。
对于单原子在Ti2CO2表面上的结合能(Eb),吸附质在催化剂表面的吸附能(Eads)将通过以下的公式计算:
式(3)中的Emetal/Mxene、EMXene和Emetal分别代表SACs的总能量、没有单原子负载的Ti2CO2体系以及单原子在相同尺寸的周期性超晶胞内的总能量。而式(4)中的Esystem、Esubstrate以及Eadsorbate则分别代表被吸附物吸附在SACs表面上的总能量、SACs的总能量以及被吸附物在相同尺寸晶胞内的总能量。
反应中的反应能垒(Ebar)和反应能(ΔE)则通过下面这个公式计算:

式中的EIS、ETS和EFS分别代表体系的初始态、过渡态以及末态时的能量。式中的ΔE结果为正值则代表反应过程是吸热反应,反之负值则为放热反应。
3 结果与讨论
3.1 单原子负载的Ti2CO2的稳定性
几何优化弛豫后的Ti2CO2的最优构型如图1所示。对于金属单原子催化剂来说,首先,选择合适的载体的先决条件是金属原子吸附在载体表面上能够以单原子形式稳定分散在载体表面,而不会形成金属团簇;其次是金属原子在载体上的结合能的绝对值要大于单个金属原子的内聚能61。图1a中所标记的数字代表单原子可能的负载位点,位点“1”指的是单原子位于三个相邻的O原子中间并在C原子的上方;位点“2”则是单原子位于单层Ti2CO2表面上的Ti原子的上方;而位于O原子上方的则是位点“3”。
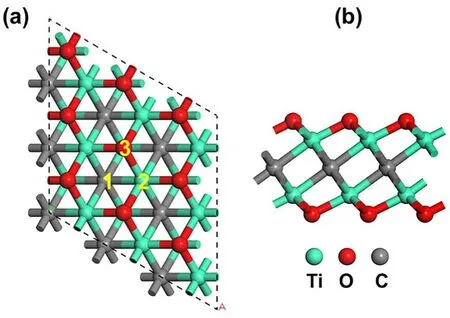
图1 单层Ti2CO2 的俯视(a)和侧视图(b)Fig. 1 Top (a) and side (b) views of the geometry of Ti2CO2 monolayer.
为了探究单原子在Ti2CO2表面上的稳定性,我们计算了Ti、V和Cr三种过渡金属在Ti2CO2表面上的结合能和Hirshfeld电荷(见表1)。结果表明:Ti和V原子最稳定的位点在C原子上方也就是位点“1”,其结合能分别是−6.99和−5.38 eV,在绝对值上比其内聚能更大;而Cr原子虽然也是在位点“1”的结合能最大(−3.88 eV),但是其绝对值比Cr原子的内聚能小(−4.10 eV),这说明Ti和V原子能稳定存在于Ti2CO2表面上,Cr原子则不能稳定存在于该MXene表面上。故而,我们接下来主要关注于Ti和V原子负载于载体表面的计算。
考虑到单原子可能在载体表面形成团簇的问题,我们进一步计算了Ti和V原子在Ti2CO2表面扩散行为。如图2所示,Ti和V原子在最稳定的位点“1”(图2中的IS)上向最近的“1”号位点(图2中的FS)扩散,中间需要跨越位点“2”(图2中的MS)。可以看出,Ti(V)原子的扩散路径中,速率限制步骤的能垒高达1.63 eV (1.10 eV),而且从IS到MS之间是一个吸热反应,反应能为0.67 eV (0.82 eV),这也就意味着不管是Ti还是V原子在Ti2CO2表面都不会发生扩散行为,从而避免了金属原子的团簇现象的发生。总之,结合能和扩散行为的结果表明Ti和V原子能以单原子形式稳定分散在Ti2CO2表面,而不会产生团簇现象。

图2 Ti (a)和V (b)原子在Ti2CO2 表面上的扩散路径图Fig. 2 The diffusion pathway of anchored Ti (a) and V (b) on Ti2CO2 monolayer.
电子结构性质能够较为完善的阐述单原子与载体之间的相互作用。因此,为了进一步了解Ti和V原子与单层Ti2CO2之间的相互作用,如图3所示,我们计算了Ti/Ti2CO2和V/Ti2CO2的差分电荷密度(DED map)和分波态密度(PDOS)。如图3a,b所示,电子都从单原子Ti(V)向基底Ti2CO2转移,然后电荷主要聚集在在Ti-O (V-O)键上,这一结果和Hirshfeld电荷分析的结果相吻合,Ti和V原子分别向载体Ti2CO2转移了0.65和0.53|e|,单原子的负载导致了Ti2CO2表面上的电荷进行重新分配(见表1)。单金属原子与基底之间的少量的电荷转移或许能提高SACs的催化活性,这与Cheng等30报道的单原子催化剂Pd/OV-Mo2CO2催化氧化CO的结果类似。

表1 3 种过渡金属在单层Ti2CO2 表面上不同修饰位点上的结合能(eV)、Hirshfeld 电荷分布(|e|)、单原子的内聚能(eV)以及电负性(eV)Table 1 The binding energies Eb (eV), Hirshfeld charge (|e|), cohesive energy (eV) and electronegativity (eV) of 3 single atoms at different decorated sites on the Ti2CO2 monolayer.
图3c是Ti/Ti2CO2中的单原子钛Ti-3d轨道和基底上与该单原子相成键的3个氧原子的O-2p轨道的PDOS图,我们可以直观的看到在能级为−6、1和3 eV左右的地方,Ti-3d轨道和O-2p轨道有较强的相互作用,这也就意味着单原子Ti和基底有较强的相互作用,单原子能稳定存在Ti2CO2表面上;V/Ti2CO2有相类似的结果,其中V-3d轨道O-2p轨道则是在−6、−0.5和3.5 eV左右(见图3d)。根据d带中心理论,金属原子的d轨道在费米能级附近的主峰与费米能级之间的距离,一般决定着吸附气体与金属原子之间的相互作用强度以及催化活性,我们发现V-3d轨道的主峰更接近费米能级。因此,V/Ti2CO2可能会与H2S分子之间有较强的相互作用,近年来的报道也提出过类似的结论29,30。

图3 单原子Ti (a)和V (b)负载在单层Ti2CO2 表面上差分电荷密度的俯视和侧视图,其中等电荷密度值为±0.032 a.u.。青色和红色分别代表电荷的聚集和丢失区域。(c) Ti/Ti2CO2 和(d) V/Ti2CO2 的分波态密度,虚线代表费米能Fig. 3 Top and side views of the deformation electron density of Ti (a) and V (b) atom anchored on Ti2CO2 monolayer with an isovalue of ±0.032 a.u., where the cyan and red regions represent electron accumulation and loss, respectively (color online). PDOS of Ti/Ti2CO2 (c) and V/Ti2CO2 (d). The pink dashed line indicates the Fermi level.
3.2 H2S在Ti/Ti2CO2和V/Ti2CO2表面上的解离
为了深入理解硫化氢分子的催化解离行为(H2S → HS* + H* → H2+ S*),本研究采取过渡态搜索方法对H2S分子在两种不同SACs表面上解离的路径进行了分析,并且绘制了相关的势能分布图(图4),H2S分子解离路径中每个状态的结构图见图5。我们首先将H2S分子分别放在Ti/Ti2CO2和V/Ti2CO2表面上进行结构优化,在考虑了H2S所有的可能的吸附构型情况下,我们发现一个非常有趣的现象,在Ti/Ti2CO2或者V/Ti2CO2表面,H2S一旦被吸附上去以后,我们首先将H2S分子分别放在Ti/Ti2CO2和V/Ti2CO2表面上进行结构优化,在考虑了H2S所有的可能的吸附构型情况下,我们发现一个非常有趣的现象,在Ti/Ti2CO2或者V/Ti2CO2表面,H2S一旦被吸附上去以后,会在催化剂表面直接解离成HS*和一个质子,其中HS*与单原子相结合,质子则与Ti2CO2表面上的O形成一个OH基团(见图5a,d),这也就意味着硫化氢解离的第一步反应(H2S → HS* + H*)没有能垒。HS*/H*的吸附能和H-S键以及S原子和金属单原子的键长的参数对比见表2。我们发现,H2S解离后形成的H-S键的键长大于H2S气体中的H-S键(0.135 nm),尤其是在V/Ti2CO2上(0.137 nm),这也就意味着H-S键在单原子催化剂V/Ti2CO2上更容易被激活,从而可能更容易断键。

表2 H2S 分子在Ti 和V 修饰的Ti2CO2 表面上自动解离后的HS*/H*的吸附能(eV),以及金属单原子与硫之间(S-Metal)的键长和H-S 键的键长(nm)Table 2 The adsorption energies Eads (eV) of HS*/H* on SACs surface, the bond length (nm) of the single atom and S atom, and the bond length (nm) between the S atom with the corresponding bonded H atom.

图4 H2S 分子在Ti/Ti2CO2 和V/Ti2CO2 表面解离的势能分布图Fig. 4 Energy profiles for H2S dissociation on the Ti/Ti2CO2 and V/Ti2CO2 surface.
如图4所示,H2S分子在Ti/Ti2CO2表面上自动解离成HS*/H*会释放1.93 eV的热量。在下一步反应中,如图5b所示,H-S键断开后解离出来的质子则和OH基团上的质子结合形成H2,剩下的S原子则和基底上的Ti相连接(图5c)。这一步反应,需要跨过一个0.86 eV的能垒,才能将H-S键断开。反观H2S分子在V/Ti2CO2表面上的情况,首先H2S发生自动解离,并释放2.26 eV的热量,相比于在Ti/Ti2CO2表面上多释放出了0.33 eV,其次H-S键断开形成H2和S*只需要跨越一个低至0.28 eV的能垒,并且释放0.23 eV的热量,这说明该反应无论是在动力学还是在热力学上面,都是较为容易进行的。总而言之,单金属原子V负载的Ti2CO2的单原子催化剂在对H2S分子的催化解离的能力比Ti/Ti2CO2更强,而且能在室温下将H2S分子有效分解成S 单质和H2。此外,使用单原子催化剂V/Ti2CO2处理H2S,整个反应的速率限制步骤的能垒相比于已经报道过的其它体系,例如:Fe/Graphene (1.95 eV)23、TiO2(1.40 eV)14、ZnO(0.94 eV)62以及Fe2O3(0.57 eV)63等许多催化剂都要低,该体系虽然和已经报道的Pt/Graphene (0.28 eV)10的能垒持平,但是由于Pt是贵金属,因为其价格原因也就限制了该体系在工业上的应用。总而言之,V/Ti2CO2分解H2S的能力可能比这些体系要高。
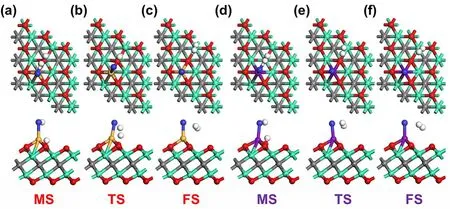
图5 H2S 分子在Ti/Ti2CO2 (a–c)和V/Ti2CO2 (d–f)表面解离路径中的各个状态的俯视和侧视图Fig. 5 Top (upper) and side (lower) views of each state of the dissociation process of a H2S molecule on Ti/Ti2CO2 (a–c) and V/Ti2CO2 (d–f) surface.
此外考虑到催化循环效应的影响,我们计算了FS态中单个S原子在单原子催化剂体系表面的吸附能,其中在Ti/Ti2CO2表面的吸附能是−1.78 eV,而在V/Ti2CO2表面上的吸附能为−1.37 eV。可以看出,S原子无论是在Ti/Ti2CO2还是在V/Ti2CO2表面上的吸附能都远低于S原子的内聚能(−2.85 eV)61,这表明S原子在两个单原子催化剂体系都能够团聚形成稳定的单质硫S8,为了反应能达到催化循环的效果,我们研究了S8在两个单原子催化剂上的吸附行为(详见图6),S8在催化剂Ti/Ti2CO2和V/Ti2CO2表面上的吸附能分别为−0.35和−0.31 eV,较弱的物理吸附表明S8在两个催化剂表面都较为容易脱附,进而完成催化循环。
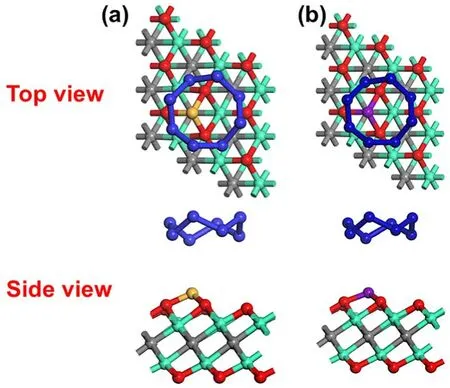
图6 S8 在Ti/Ti2CO2 (a)和V/Ti2CO2 (b)表面吸附的主视和侧视图Fig. 6 The top and side views of S8 adsorbed on the Ti/Ti2CO2 (a) and V/Ti2CO2 (b) surface, respectively.
根据先前的报道32,64,只有当反应能垒小于0.91 eV的临界势垒时,反应才会在室温下发生。而本工作中,H2S在V/Ti2CO2上分解反应的能垒只有0.28 eV,所以该反应在室温下可以发生。考虑到在化工行业中,催化剂在高温条件下处理硫化氢的操作温度一般在353-523 K18,为了进一步了解获H2S分子在实际的条件下的分解行为,我们根据下列公式计算了H2S在V/Ti2CO2上分解反应的时间28,32:

式(7)中A代表震动频率(1012Hz),Ebar则代表反应能垒,此处反应能垒为0.28 eV,kB代表玻尔兹曼常数(8.63 × 10−5eV·K−1),T代表温度。由此计算出来在T= 500 K时,反应时间为65.79 ns,在常温下(T= 300 K),反应时间为4976.10 ns。因此,H2S无论是在常温下还是在实际操作温度下,在V/Ti2CO2表面上的上分解反应具有非常快的反应动力学。而且高温还会加快反应速率,比常温下将近快了两个数量级。
为进一步了解单原子与H-S键之间的界面键合作用,我们对HS*吸附在两种催化剂表面的原子进行了PDOS分析,如图7所示。首先,我们可以看出H2S分子中的H-S键的成键轨道(图7a)主要是由位于价带中的H-1s和S-3p轨道的相互的杂化作用而贡献的。图7b中显示的是HS*吸附在Ti/Ti2CO2的PDOS图,从图中可以看出,在单原子Ti的作用下,无论是H-1s还是S-3p轨道相比气体中都生向左偏移大约1 eV。而图7c中,H-1s还是S-3p轨道相比吸附之前,向左偏移了大约3 eV,不仅如此,相比于图3d,V原子在HS*吸附的影响下,V-3d轨道在费米能级附近多产生了1个峰,表明V原子与H-S键的相互作用很强,而且,费米能级附近的金属峰能够激活H-S键,从而较为容易地将HS*分解成S单质,这与过渡态搜索的结果一致。总的来说,相比于Ti/Ti2CO2,单原子催化剂V/Ti2CO2更能将H-S键断开,从而有利于H2S分子的整体解离过程的进行。
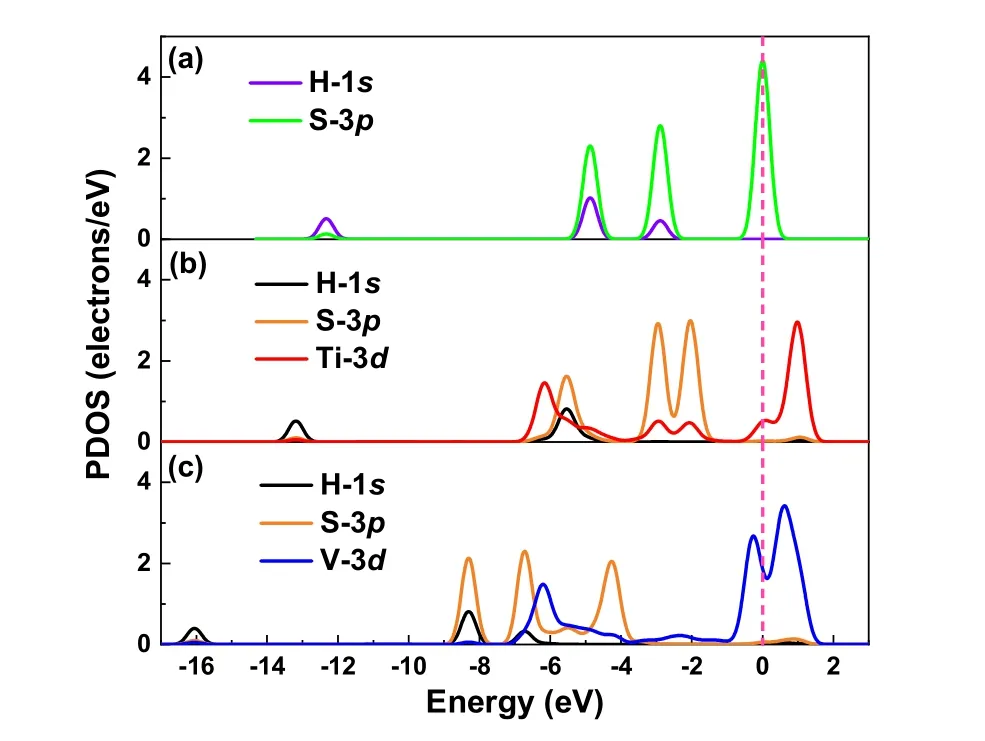
图7 H2S 分子在Ti/Ti2CO2 (b)和V/Ti2CO2 (c)表面吸附前(a)后(b,c)的S、H 以及单原子的PDOS 图Fig. 7 The PDOS of S, H and single atom before (a) and after adsorbed on Ti/Ti2CO2 (b) and V/Ti2CO2 (c) surface.
4 结论
在本工作中,我们采用密度泛函理论研究了单金属原子Ti、V和Cr三种过渡金属负载Ti2CO2的单原子催化剂的稳定性。结果表明,Cr金属因为在Ti2CO2表面的稳定性较差,而Ti和V两种金属与载体之间的结合能较大,不仅能以单原子形式稳定分散在Ti2CO2表面,而且不会产生团簇现象,从而能够形成稳定的单原子催化剂。Hirshfeld电荷分布和PDOS结果表明,单原子的负载导致了Ti2CO2表面上的电荷进行重新分配,V/Ti2CO2可能会与H2S分子之间有更强的相互作用。接下来我们考察了两种不同金属原子负载Ti2CO2,对硫化氢解离的差异。结果表明:两种原子单独负载的单层Ti2CO2都能够让H2S分子发生自动解离生成-HS和一个质子(HS*/H*),但是V/Ti2CO2中的V原子能激活H-S键,并将其分解,在跨越一个很小的能垒(0.28 eV)之后,该单原子催化剂在室温下就能将H2S转化成硫原子和氢气,而且S原子在单原子催化剂V/Ti2CO2表面能够团聚形成稳定的单质硫,从而完成催化循环。此外,在500 K下,H2S在V/Ti2CO2上分解反应的时间为65.79 ns,反应时间比常温下将近快了两个数量级。除此之外,在使用该SACs分解H2S,整个反应的速率限制步骤的能垒相比于已经报道过的其它体系都要低,而且该体系在经济可行性和处理能力方面都有较大的潜力。综上,本研究为开发高效硫化氢去除的催化剂以及扩大MXene基材料应用范围提供重要指导作用。
猜你喜欢
杂志排行

物理化学学报的其它文章
- Defect Engineering in Two-Dimensional Graphitic Carbon Nitride and Application to Photocatalytic Air Purification
- 理论计算评价光催化剂VOCs 降解性能:g-C3N4 量子点/石墨烯
- 富含缺陷的2D/2D 异质结促进光催化清洁能源转化
- Development of Iron-Based Heterogeneous Cocatalysts for Photoelectrochemical Water Oxidation
- 二维光催化材料电子结构和性能调控策略研究进展
- Non-Noble-Metallic Cocatalyst Ni2P Nanoparticles Modified Graphite-Like Carbonitride with Enhanced Photocatalytic Hydrogen Evolution under Visible Light Irradiation