1 例反复脑卒中发作Fabry 病患者的临床资料及家系基因检测结果分析
2021-08-25陈林发李友戴永武贺文麟刘娟丽蔡秀曲黄靓唐劭年李江黄志勇颜津津
陈林发,李友,戴永武,贺文麟,刘娟丽,蔡秀曲,黄靓,唐劭年,李江,黄志勇,颜津津
1 广州医科大学惠州医院惠州市第三人民医院神经内科,广东惠州516002;2 广东医科大学附属医院广东省衰老相关心脑疾病重点实验室
Fabry 病是X 染色体连锁遗传代谢疾病,由GLA 基因突变导致体内的α-半乳糖苷酶A(α-GalA)活性部分或全部缺失,使其底物酰基鞘氨醇己三糖(GB3)无法被降解而蓄积在全身的血管与组织内,对脏器的结构及功能造成不可逆的损害[1]。缺血性脑卒中是Fabry 病的常见并发症,在对5 978 例以脑卒中为表现的Fabry 病高危患者筛查中发现,GLA基因突变的男性占0.13%、女性占0.14%,其中诊断为Fabry 病患者中,急性脑血管事件发生率为13%,其中男性占15%、女性占11.5%[2]。2016年,临床发现1 例青年脑卒中患者,3年内先后3 次出现后循环脑梗死症状,结合其他临床症状及基因检测结果最终确诊为Fabry 病,现对其临床资料及其家系基因检测结果进行分析,以提高对Fabry 病的认知及诊治水平。
1 资料分析
1.1 临床资料 患者陈某,男性,于2016年12月(34 岁)因反复发作性视物重影1 个月,伴右侧肢体麻木无力第一次就诊于惠州市第三人民医院,头颅MR 检查提示右侧桥臂脑梗死,予阿司匹林及立普妥常规治疗,出院后长期规律服用。患者于2019年1月突发眩晕、右侧肢体麻木无力,伴恶心呕吐及视物重影第二次入住我院。查体显示构音欠清,右侧上下肢轻瘫试验阳性,右侧偏身浅感觉减退,右侧Babinski 征阳性、Chaddock 阳性,NIHSS 评分2 分。实验室检查结果显示,尿微量白蛋白148.00 mg/L,尿肌酐20.3 mmol/L,24小时尿蛋白定量0.20 g/24 h,总胆固醇3.50 mmol/L,低密度脂蛋白胆固醇1.64 mmol/L,胸部DR 检查显示患者心影增大,右心声学造影提示卵圆孔未闭、右向左分流,头颈CTA检查提示双侧小脑半球、丘脑及基底节对称性钙化,椎基底动脉扩张。结合临床表现及相关检查结果,考虑患者为Fabry 病相关性卒中、椎基底动脉异常扩张、卵圆孔未闭。患者发病4.5 h 内即到院治疗,在静脉溶栓时间窗内,治疗上予阿替普酶60 mg 静脉溶栓,复查头部MR 提示脑桥左份多发亚急性小梗死灶,患者出院后予华法林、立普妥治疗,长期随访患者INR 均能控制在2.0~3.0。患者出院2月后至北京某医院进一步完善相关检查,结果显示神经传导速度、F 波、H 反射、视频眼震电图及脑血液动力学未见异常,皮肤交感反射提示双掌心交感皮肤反应START. N1. P1 均延长,双足心交感皮肤反应START.N1.P1 均未引出,听觉检查提示听力减退、耳鸣,视觉检查考虑Fabry相关眼部改变。Lyso-GL-3检测为88.41ng/ml;Lyso-GB3检测为165 ng/ml;α-GalA检测为0.5 nmol/(g·min)。患者于2019年8月突发言语不清、行走不稳、头晕及视物重影第三次入住我院。入院查体显示言语稍不流利,右侧偏身麻木感,左侧共济征稍差,双侧巴氏征(-);复查颅脑MR 提示左侧桥臂急性脑梗死,予抗凝、降脂治疗。2020年5月,患者至广州某医院复查,换用西洛他唑及立普妥治疗至今。
1.2 既往史 患者3 岁时出现无明显诱因无汗、怕热,夏天时害怕出门;8 岁时出现双侧手脚疼痛,双足趾明显,休息后症状可稍缓解,曾多次就诊于当地医院,考虑末梢神经炎、雷诺病,治疗效果不佳。患者初中时肢端疼痛逐渐加重,约23岁时双侧大腿根部及四肢末端开始出现散在的1~5 mm大小暗红色或紫红色圆形丘疹,近4年受凉后易出现腹泻。
1.3 家系调查 患者女儿约6 岁时发现平时活动及出汗较同龄孩子少,怕热,常诉腹痛及肢端末梢疼痛,查体可见皮肤血管角质瘤,纯音测听提示双耳高频听力下降,发泡实验阴性,Lyso-GL-3 检测为82.95 ng/ml;Lyso-GB3检测为137 ng/ml。患者母亲于2011年(54岁)因脑出血至行钻颅减压手术,遗留右侧肢体麻木症状,查体显示右侧肢体肌力5-级及感觉减退,右测Babinski 征阳性,尿微量白蛋白稍升高,胆固醇5.82 mmol/L,低密度脂蛋白2.85 mmol/L,Lyso-GL-3检测为9.32 ng/ml;心电图检查提示ST段缺血型改变及T波变化,左心室高电压,彩超提示左室肥厚。
1.4 基因检测 用抗凝管抽取患者、患者女儿、患者母亲外周静脉血各2 mL,采用DNA提取试剂盒提取基因组DNA,并测量其吸光度值及浓度,由广州金域公司通过对4 811 个临床相关基因的外显子区进行高通量测序,结合患者临床资料和生物信息学软件预测结果筛选突变基因,并进行Sanger 测序验证。基因检测结果显示,患者、患者母亲、患者女儿均存在GLA 基因g.8319 A>G 突变(图1),为X 染色体连锁遗传代谢疾病,符合Fabry病诊断。
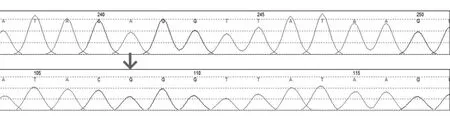
图1 患者、患者母亲、患者女儿基因检测结果(突变位点A>G)
2 讨论
Fabry 病又称糖鞘脂类沉积症,1898年由英格兰的安德森和德国的FABRY 几乎同时报道。早在2011年YOSHIMITSU 等[3]就 报 道 过 该 突 变 位 点g. 8319 A>G,由于该内含子区剪接位点发生变化,影响了GLA基因转录,导致整个外显子4缺失,最终使得所编码的蛋白质丧失其正常功能,为很强的致病性变异[4]。本研究中的患者是一个具有典型Fab⁃ry 病特征的家族,该家系中患者、患者母亲、患者女儿均携带GLA 基因g. 8319 A>G 突变。YOSHIMIT⁃SU 等[3]报道该基因突变位点与心血管事件相关性较大,然而该患者及患者母亲却均发生了脑卒中,因此,该突变位点是否可预测脑卒中发生,有待进一步研究验证。
约30%Fabry女性患者酶的活性在正常范围,因此女性携带者所表现出的临床症状较轻,寿命甚至可以正常。在该家族中,患者母亲为女性杂合子迟发型病例,各临床症状均较患者轻,虽已累及泌尿系(尿微量蛋白升高)、心脏(心室肥厚、左室高电压)、听觉及视觉系统(听力、视力稍下降)及中枢神经系统(卒中),但却没有典型的皮肤血管角质瘤、少汗及胃肠道症状。而先证者为经典型Fabry 病,其3岁时即出现神经系统病变,累及周围神经,出现典型的肢端感觉异常和发作性疼痛,紧张、运动、冷热等刺激时可诱发疼痛,随年龄增长疼痛逐渐减轻;自主神经受累则表现为皮肤常干燥、少汗,检查双掌皮肤交感反射延长及双足皮肤交感反射未引出等。患者皮肤病变虽未见典型血管角质瘤分布于大腿根部及坐浴区,但是手指末端及背部却可见多发直径2~4 mm暗红色毛细血管扩张性斑点或丘疹。同时,患者青年时即出现Fabry 相关性眼部病变,导致视物模糊及视力下降,同时也出现听力减退及耳鸣症状。患者在肾早期损伤指标中出现轻微蛋白尿,这是患者慢性肾损伤的结果。患者胸部DR 检查提示心脏增大,虽彩超未见明显心功能异常,但患者近年已出现无明显诱因的胸闷不适症状,应该密切监测患者心功能情况。
双侧对称的纹状体-苍白球-齿状核钙化又被称为Fahr’s 综合征,临床表现从无症状到各种运动障碍及神经精神表现等均有报道[5]。本研究中患者头部CT提示双侧小脑齿状核、丘脑及基底节对称性钙化,患者母亲头部CT 提示双侧小脑半球、双侧基底节、右侧额叶白质区、右侧放射冠及左侧枕叶皮层多发钙化灶。目前认为丘脑枕钙化为Fabry 病影像学特征性改变[6],然而该患者中枢神经系统病变为多发对称性钙化,以双侧苍白球、双侧丘脑、小脑为主,且范围较正常明显增大,考虑病理性病变,这是既往Fabry 病中尚未报到过的影像学表现。因此我们考虑患者、患者母亲颅内多发异常钙化是由Fabry 病所致继发性Fahr’s综合征。
Fabry病常见的死亡原因是卒中,具有高发和早发两大特点。Fabry 病卒中患者中88%的男性和59%的女性年龄≤55 岁,故中青年患者居多。有调查[7]显示,与普通人群比较,年龄在25~44岁的Fab⁃ry 病患者卒中发生率升高了12 倍,且主要为缺血性卒中,而出血性卒中相对少见。Fabry病卒中患者常表现为后循环缺血症状,基底动脉扩张是Fabry 脑血管病变的一大特征,可能与α-半乳糖苷酶的作用底物GB3选择性累及椎基底动脉系统有关。本研究中,患者反复多次发生后循环脑梗死,该患者基底动脉管径增粗,走行笔直,其中最宽管径位于基底动脉起始段,考虑患者椎基底动脉异常扩张。此外,该患者头颈CTA 提示动脉粥样硬化并不明显,我们推测慢性扩张的基底动脉引起血管储备能力下降,导致脑血流局部自主调节功能失调,可能是造成脑缺血性事件发生的重要诱因。有研究[8]发现,血管舒缩运动功能及自主调节能力障碍在该病的脑血管病变中可能起到一定的作用,推测选择性后循环血管受累,与其自主神经密度少、血管自主调节能力相对较差有关。Fabry 病特征性的皮肤血管角质瘤也证实皮肤微小血管系统存在普遍的扩张。MOORE 等[9]通过正电子发射体层摄影观察到,Fabry病患者在静息状态下,后循环相对脑血流量较正常人显著增加,提示Fabry 病早期小动脉血管内皮及平滑肌细胞功能失调。同时,呼吸抑制试验检测脑血管反应性和脑血流自主调节能力结果显示,患者脑小血管CO2反应性良好,直立倾斜试验阴性,提示患者后循环小血管对二氧化碳刺激变化的反应性正常,颅内小血管调节及储备功能正常。因此,我们考虑患者基底动脉大血管为主要病变基础,脑桥旁正中动脉、桥脑短旋及长旋动脉终末端血流紊乱为结果的小血管闭塞事件,同时患者视力模糊、眩晕、耳鸣、听力下降均可能与上述动脉缺血相关,这一结果与Fabry 病的临床表现和脑血管异常具有一致性。我们认为椎基底动脉的异常扩张是该疾病的一种特别严重的表现,可能与该家族中基因的突变有关。
卵圆孔为胎儿期血液循环的生理通道,一般在出生后1年内关闭,若在3 岁后仍未闭合,则遗留永久性的缺损。卵圆孔未闭患者的血栓主要来源于静脉系统,血栓可在右心房压力增大的瞬间流入左心房而进入动脉,最终导致血栓栓塞型脑梗死。有研究表明,GB3在血液中浓度增高,类似于高同型半胱氨酸血症,可促进高凝、易栓状态的形成,而卵圆孔未闭及高凝、易栓状态下易导致青年人反复卒中。在本研究中,患者在静息状态下左房见散在气泡回升,咳嗽及Valsalva 动作后左房内气泡回声明显增多,我们确定了患者有卵圆孔未闭及右向左分流现象,这也让我们进一步思考脑梗死的病因。我们完善了血管炎、风湿、免疫、蛋白C 和蛋白S 未见血管炎及血液高凝状态,双下肢静脉彩超、心脏彩超均未见血栓。但是无创检查阴性并不能完全排除静脉源性血栓,因为大部分静脉血栓位于表浅或小且深部,需通过有创静脉造影才能发现,患者反复多次发生脑卒中,未找到静脉血栓证据,我们考虑患者反复卒中由卵圆孔未闭所致可能性较小。在第二次脑梗死诊治过程时,我们曾建议患者行卵圆孔封闭手术,患者及家属表示暂不考虑,要求抗凝保守治疗,目前Fabry 病合并卵圆孔未闭的诊治探索,在Fabry 青年卒中中也尚未报道。
患者于2016年12月第一次因脑梗死住院,出院后遵嘱规律服用阿司匹林及立普妥。在长达2年的抗血小板及降脂治疗方案后,患者于2019年2月再发脑梗死,同时也明确了Fabry 病及卵圆孔未闭的诊断。为预防卵圆孔未闭导致脑栓塞的发生,我们尝试予抗凝治疗。患者遵医嘱监测INR 均能达标,但却在2019年8月患者发生了第三次脑梗死。患者先后使用阿司匹林、华法林进行抗血小板及抗凝治疗,但效果欠佳。目前尚无Fabry 病患者进行抗栓及抗凝治疗能有效预防卒中的数据,结合患者前后使用抗血小板及抗凝等规律治疗方案并达到治疗标准条件下仍发生卒中,提示Fabry 病相关性卒中进行常规抗血小板、抗凝治疗效果不甚理想。阿替普酶用于急性缺血性脑卒中合并Fabry 病的治疗报道非常有限,KARGIOTIS曾报道过1例Fabry患者在急性缺血性脑卒中使用阿替普酶,患者NIHSS 评分从入院2分,到出院临床症状改善,NIHSS评分1分[10]。在第二次卒中时,该患者在4.5 h 内到院,我们按照2018年卒中指南予阿替普酶0.9 mg/kg 静脉溶栓,Fabry 病所致的脑卒中静脉溶栓获得了良好的效果并未出现明显并发症,出院时患者症状稍好转。该病例也进一步支持了Fabry 病患者在急性缺血性脑卒中静脉溶栓是安全的,弥补了罕见病缺血性卒中静脉溶栓临床数据的不足。
综上,我们首次报道了GLA g.8319 A>G突变与Fabry基底动脉扩张所致后循环梗塞的相关,首次描述了Fabry 病家系颅内多发异常钙化的影像学表现,同时分析了Fabry 病、基底动脉异常扩张、卵圆孔未闭的青年脑卒中患者的临床表现及在抗血小板、抗凝、静脉溶栓等多种常规卒中诊治方案。我们认为抗血小板及抗凝方案均不是Fabry 患者脑卒中有效预防方案,静脉溶栓治疗并非Fabry 病急性缺血性脑卒的禁忌症。
