协同碱金属和碱土金属卤化物催化CO2和环氧化合物 环加成反应的助催化剂研究进展
2021-08-19李庆硕李梦情赵馨蕊赵可新李双儿常海波
李庆硕, 李梦情, 赵馨蕊,赵可新,李双儿,常海波
(河南大学 化学化工学院, 功能聚合物复合材料研究所, 河南 开封 475004)
CO2是主要的温室气体之一,同时也是廉价易得、无毒、可再生的C1资源。随着环境和能源问题的日益严峻,CO2的资源化利用受到了人们越来越多的关注[1-5],其中将CO2与环氧化合物环加成转化为环状碳酸酯(图1)是CO2资源化利用中最有效的路线之一[3-4]。这不仅因为该反应是典型的原子经济反应,有100%的原子利用率,而且反应所得产物环状碳酸酯具有高沸点、高溶解性和可降解等一系列优异性能,广泛应用于有机合成、电化学、纺织和聚合物合成等领域[6]。

图1 CO2与环氧化合物的环加成反应Fig.1 Cycloaddition reaction of CO2 and epoxides
在CO2和环氧化合物合成环状碳酸酯的反应中,环氧化合物具有较高的环张力和很高的反应活性,易发生开环加成反应;然而CO2中碳原子处于最高氧化态,具有热力学上的稳定性和动力学上的惰性。因此一般要在催化剂的作用下该反应才能够顺利进行。据报道碱金属卤化物[7-8]、碱土金属卤化物[9]、金属氧化物[10]、过渡金属配合物[11-14]、金属有机框架[15-17]和有机催化剂[18-20]等均可以作为催化剂促进CO2与环氧化物的环加成反应。
碱金属和碱土金属卤化物因低毒、成分简单、易处理、廉价易得且稳定而受到广泛地研究,尤其是碱金属卤化物催化剂目前已经应用于工业化生产。然而,这两类金属卤化物单独做催化剂,催化活性均比较低,需要在助催化剂的作用下才表现出较高的催化活性。目前已经开发出各种类型的助催化剂可以有效地协同碱金属和碱土金属卤化物催化CO2与环氧化物的环加成反应。
本文对这两类金属卤化物的助催化剂进行综述,以促进其在CO2资源化领域的开发和应用。
1 碱金属卤化物催化体系
对CO2和环氧化合物环加成反应的机理研究表明,该反应的历程分为三步,即环氧化合物开环,二氧化碳插入,然后分子内闭环。其中环氧化合物的开环是关键步骤。常见的措施有两种:
第一,加入亲核性试剂促进其开环。卤素阴离子是比较常用的亲核性基团,尤其是碘负离子(I-),它具有比较强的亲核性且在分子内闭环时比较容易离去,在该类反应中最为常用。对于碱金属卤化物而言,正负离子之间存在强的离子键作用,这一方面使卤素阴离子的进攻能力较弱,另一方面使其在环氧化合物中的溶解度比较低,因此单独使用,其催化活性比较低。引入相转移助催化剂以提高碱金属卤化物的催化活性是有效方式之一。相转移催化剂一方面可以增加其溶解度,另一方面可以减小阴阳离子的相互作用,从而增强卤离子的亲核进攻能力。
第二,路易斯酸活化环氧化合物。环氧化合物中的氧带有孤对电子,易与路易斯酸(通常是氢键供体HBD或金属离子)作用,极化三元环中的碳氧键,从而活化环氧化合物,当其受到亲核试剂进攻时,更容易开环。碱金属离子与环氧化合物中的氧之间也存在一定的静电作用,但碱金属离子的电荷为+1,离子半径一般比较大,因此作用比较弱,不能使环氧化合物有效地活化,需要引入路易斯酸化合物对环氧化合物进行活化。此外,助催化剂中若含有可以活化CO2的含氮基团,则可进一步降低活化能,促进反应进行。经过多年的研究,目前已经开发出来了多种助催化剂。
本文将助催化剂分为四类进行综述,即氢键供体类(HBD)、路易斯酸类、配体化合物类、含有HBD的配位类等。
1.1 氢键供体类助催化剂
2003年,SHI等[21]发现NaI/PPh3/PhOH物质的量之比为1/1/1,浓度为2.0%(摩尔分数),4 MPa和120 ℃条件下,反应4 h,产率可达100 %。苯酚在其中扮演着重要作用,他们认为苯酚中的酚羟基作为HBD与环氧丙烷(PO)中的氧形成氢键作用而使碳氧键极化,进而起到活化PO和促进了反应进行的作用(图2)。

图2 NaI/PPh3/PhOH催化二氧化碳和环氧丙烷环加成反应机制[21]Fig.2 Plausible reaction mechanism for CO2-PO cycloaddition reaction catalyzed by NaI/PPh3/PhOH[21]
酚中酚羟基的位置和数量与其催化活性密切相关。JI等[22]的研究发现含有多酚羟基的天然产物单宁酸(TA)与KI组成的二元催化体系可以在使用较少量的催化剂(1% KI和0.25% TA)和较为温和的条件(100 ℃和1.0 MPa)下催化氧化苯乙烯(SO)和CO2的环加成反应,反应16 h,产率为93%。为阐述其中的原因,作者进一步研究了邻苯二酚、对苯二酚和1, 2, 3-苯三酚的催化活性,发现顺序为:1, 2, 3-苯三酚>邻苯二酚>对苯二酚。他们认为苯环中的相邻酚羟基不仅易与环氧化合物形成氢键,有利于其开环,而且能更好地稳定开环所形成中间体,从而有利于环加成反应的进行。
除酚羟基外,研究人员还发现醇羟基也可以协同KI催化该类反应。如β-环糊精(β-CD)[23]、天然纤维素[24]、生物质废弃物甘蔗渣[25]和聚乙烯醇[26]均可以与KI一起有效地催化CO2和环氧化合物的环加成反应。然而,与酚类化合物相比,醇羟基类的助催化剂用量较大,反应条件也较为苛刻。如采用β-CD作为助催化剂时,催化剂的用量为2.5% KI和0.1 gβ-CD,反应要在6 MPa和120 ℃的苛刻条件下进行。这可能与其酸度系数(pKa)有关。与酚羟基相比,醇羟基的pKa较大,其与环氧化合物中的氧形成的氢键较弱,对碳氧键的活化作用有限,苛刻条件下才能使环氧开环。
2013年,CHENG等[27]报道了三乙醇胺(TEA)(图3)与KI协同可以在110 ℃和2 MPa条件下催化该反应。当用乙醇或三乙胺代替TEA时,同等条件下,PO的转化率从91%下降到了5%和25%。他们认为TEA表现出较好催化活性的原因是由于其上除了带有可以活化PO的羟基外,TEA中的N原子还可以与CO2形成氨基甲酸盐,起到活化CO2的作用。基于这一认识,类似的催化体系如氨基醇[28]、氨基酸[29-33]、聚多巴胺球[34]和羊毛粉[35]等与KI组成的协同催化体系先后被报道可以在相对温和的条件下催化该类反应,其中聚多巴胺球和羊毛粉与KI组成的催化体系具有较好的重复利用性。

图3 三乙醇胺-碘化钾协同催化二氧化碳和环氧丙烷环加成反应机制[27]Fig.3 Possible mechanism for CO2-PO cycloaddition reaction catalyzed by triethanolamine/KI [27]
近年来,人们也发现一些含HBD的无机材料如Fe3O4@Fe(OH)3核壳复合材料[36]、富勒烯醇[37]等也可以用作助催化剂协同KI催化该类反应,反应机制类似。与含HBD的有机类助催化剂相比,这些助催化剂更加稳定,具有更好的重复利用性,如富勒烯醇和KI催化体系,重复利用十次,催化活性不降低。
总之,HBD类化合物均可不同程度地提高碱金属卤化物的催化活性,当该类化合物中含有能够活化CO2的功能基团时,能进一步增强催化体系的催化活性。但HBD化合物中HBD的数量和pKa的大小和催化体系的关系还有待进一步研究建立,在温和条件下协同碱金属卤化物催化该类反应的HBD类助催化剂亟待开发。此外,目前开发的HBD类助催化剂主要集中在羟基类化合物,而其它化合物研究较少。
1.2 路易斯酸类助催化剂
除HBD外,金属阳离子和非金属阳离子均可以与环氧化合物中的氧相互作用而使碳氧键极化,进而起到活化环氧化合物的作用。尤其是金属阳离子,其上的空轨道可以和环氧化合物中的氧产生配位作用而使其活化。2011年HAN课题组[38]采用金属有机框架材料MOF-5作为助催化剂协同KI催化该反应,取得了一定的催化效果,MOF-5中Zn4O团簇作为Lewis酸性中心与环氧环上氧原子配位,KI中的I-进攻环氧环上位阻较小的β碳原子引发环氧烷烃开环。但该催化体系中KI的用量较多(2.5%),反应条件比较苛刻(6 MPa、90 ℃和2 h)。WANG等[39]报道了二氯二茂钛(Cp2TiCl2)协同KI催化该类反应的情况。在150 ℃和1.2 MPa条件下,采用2%的KI和1%的Cp2TiCl2,反应4 h,PC产率为98%。他们认为Cp2TiCl2首先与KI作用形成了Cp2TiI2,该化合物中的Ti2+与PO中的氧产生配位作用,从而使其活化(图4)。

图4 二氯二茂钛-碘化钾协同催化反应机制[39]Fig.4 Proposed mechanism for CO2-epoxide cycloaddition catalyzed by titanocene dichloride/KI [39]
同时含有金属离子和HBD的化合物也有报道。如YU等[40]以钛酸盐纳米管(TNT)做助催化剂。钛酸盐纳米管中的Ti4+可以作为路易斯酸活化环氧化合物,而且钛酸盐纳米管表面富含羟基,可以HBD活化环氧化合物。在120 ℃和3 MPa条件下,使用5.0% KI和0.1 g TNT,反应6 h,PC产率为84.1%。但反应条件较为苛刻,而且反应过程中需要用到有机溶剂二氯甲烷。

图5 BPO4-KI催化CO2和环氧化合物环加成反应的机制[41]Fig.5 Possible reaction mechanism for cycloaddition of CO2 with epoxides over BPO4/KI[41]
最近,MU等[41]报道了含有非金属路易斯酸活性位点的硼磷酸(BPO4)协同KI催化的反应,在无溶剂条件下,110 ℃和4 MPa,反应6 h,PC产率为98%。而且该催化剂比较稳定,循环使用五次,产率几乎不变。该体系中,BPO4中的硼羟基和磷阳离子分别作为HBD和路易斯酸活性位点与环氧化合物作用而使其活化。
路易斯酸类助催化剂稳定性好且与反应物和产物均不相容,容易与产物分离,一般具有重复使用性能好的特点。然而相比HBD反应体系,目前开发的路易斯酸类助催化剂对环氧化合物的活化能力有限,因此反应条件苛刻,有些甚至需要有机溶剂。通过调节配位阴离子来调控其与环氧化合物的作用,开发出温和条件下的路易斯酸类助催化剂是未来值得探究的方向。
1.3 配体类助催化剂
碱金属卤化物在环氧化合物中的溶解性低是其催化活性差的主要原因之一。冠醚可以作为相转移剂,通过与碱金属阳离子形成配合物,从而提高碱金属卤化物的催化活性。1984年,ROKICKI[42]将14-冠-5和18-冠-6冠醚等相转移剂引入反应体系。研究结果表明,冠醚与碱金属离子的配位作用,增加了碱金属盐的溶解度,同时降低了阴阳离子之间的相互作用,提高了卤素阴离子的亲核性,进而提高了碱金属卤化物的催化活性。1993年,TAKESHI等[43]用苯并14-冠-5为相转移催化剂,详细研究了各种碱金属盐的催化活性。结果表明在0.1 MPa下,碱金属卤化物就具有好的催化活性,而且催化活性与CO2压力无关。但是小分子冠醚价格昂贵,不能循环使用,而且有一定的毒性,因此限制了其应用。
WERNER等[44]用聚[(二苯并-18-冠-6)](poly18C6)代替小分子冠醚,并将KI负载其上得KI@poly18C6催化剂。用2%的负载催化剂,在100 ℃和1 MPa条件下,反应3 h,产率为91%。该催化剂具有一定的重复使用性,但存在因KI的渗出导致其活性下降的问题。
除了冠醚类助催化剂外,2012年HAN等[45]报道了葫芦脲(CB[6])协同卤化钾催化CO2-PO环加成反应的反应。采用0.1 g CB[6]和1.5%的KI,在120 ℃和4 MPa条件下,反应2 h,产率可达98%,而且该催化剂重复使用五次,产率几乎没有变化。进一步的研究表明,CB[6]中的羰基与K+的配位作用是造成该催化体系活性高的主要原因(图6)。

图6 葫芦脲-碘化钾催化二氧化碳和环氧化合物环加成反应可能的机制[45]Fig.6 Possible mechanism of the coupling of CO2 with epoxide using KI/CB[6] [45]
从上述的研究可以看出,配体类助催化剂一般是通过相转移和减少阴阳离子间的相互作用,来提高卤素负离子的亲核性。然而这些助催化剂合成复杂,价格昂贵,有些甚至毒性较大,限制了其推广应用。
1.4 含有HBD的配位类助催化剂
如果助催化剂能够与碱金属卤化物中的金属离子产生配位作用,起到活化碱金属卤化物的作用,同时又含有可以活化反应底物的HBD,可以预想该催化体系将会有比较好的催化活性。
2013年,ZHOU等[46]就将季戊四醇(PER)与KI结合催化该类反应,PER中的氧原子可以与钾离子产生配位作用,对KI起到活化作用,而其中的羟基扮演HBD的作用。研究结果表明在2.5 MPa和130 ℃条件下,反应2 h,PC的产率可达98.8%。
ZHU和SCHAFFNER采用廉价易得的聚乙二醇(PEG)与KI复合催化该反应[47-48],也取得了不错的催化效果。催化效果与PEG的分子量密切相关,PEG400的催化效果最好。但该催化体系的反应条件苛刻,需要在2 MPa和150 ℃条件下,反应5 h,才能获得比较好的产率。KUMAR等[49]进一步研究发现了α,ω-羟基遥爪型聚乙二醇(PEG400)(图7)与KBr进行螯合,在温和条件下(60 ℃和0.1 MPa),催化CO2和PO的反应2 h,PC产率可达98%。聚乙二醇廉价易得且无毒,该复合催化剂制备简单,重复使用六次产率没有明显变化,但用量比较大(10%)。
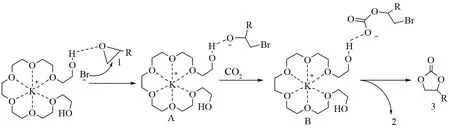
图7 α,ω-羟基遥爪型聚乙二醇-溴化钾催化反应可能的机制[49]Fig.7 Possible mechanistic pathway for cycloaddition of CO2 with epoxides over BPO4/KI[49]
类似于α,ω-羟基遥爪聚乙二醇,KANEKO等[50]采用乙二醇的齐聚物-四甘醇协同KI催化。采用10% KI和10% 四甘醇,在40 ℃和常压条件下,催化CO2和低活性底物氧化苯乙烯偶合,反应24 h,产率可达99%。通过与乙二醇、三甘醇、四甘醇二甲醚和四甘醇单甲醚的对比研究,证实了分子链的大小和四甘醇中两个羟基对催化剂活性都有重要影响。
除了乙二醇类齐聚物,LI等报道了琥珀酰亚胺(SI)也具有类似的作用[51]。SI的“NH”质子作为HBD可活化环氧化合物,而且SI存在三种共振稳定结构(如图8),其中氧负离子因与钾离子存在强相互作用,可使KI在PO的溶解度提高376.5%,起到类似配体的作用。研究结果表明5%SI和5%的KI在0.4 MPa和70 ℃条件下催化CO2和PO环加成,反应4 h,产率为97.5%。

图8 碘化钾和琥珀酰亚胺之间的作用[51]Fig.8 Interaction between KI and SI [51]
含HBD的配位类助催化剂融合了配体化合物和HBD类助催化剂的优势,在与K+配位提高I-亲核性的同时,通过氢键极化环氧烷烃的C-O键,活化环氧烷烃,促进其开环。目前,含有这类助催化剂的反应条件最为温和,但是普遍还存在催化剂用量大,反应时间比较长的缺点。此外,目前开发的这类催化体系在产物中具有一定的溶解度,需要经过减压蒸馏或者添加沉淀剂实现其重复使用,这无疑会增加能耗和产品成本,不利于其工业化应用。因此,通过固载等手段使其变成非均相催化剂将有利于其工业化应用。
2 碱土金属卤化物催化剂体系
与碱金属卤化物相比,碱土金属尤其是钙卤化物用于催化二氧化碳和环氧化合物的环加成反应近年来才受到人们的广泛关注。
碱土金属离子带有两个正电荷,自身具有活化环氧化合物的功能。但同样由于碱金属卤化物中阴阳离子相互作用强,使碱土金属离子活性环氧化合物的作用和卤离子的亲核性均比较弱,因此单独使用时催化活性也比较低。
类似提高碱金属卤化物催化活性的策略,采用相转移催化剂是提高碱土金属卤化物催化活性的有效途径之一。2003年,KOSSEV等[52]尝试用相转移剂季铵盐或季膦盐提高氯化钙的催化活性,但是反应条件苛刻,在170 ℃和4 MPa条件才具有一定的活性。在随后十几年中,人们对这类催化剂的关注较少。2017年,WERNER等[9]发现在18-冠-6相转移催化剂存在下CaI2可以在温和的条件(甚至在室温23 ℃和1 atm)下有效地催化CO2与各种类型环氧化合物的环加成反应。如在23 ℃和1 atm条件下,采用5% CaI2和5%的18-冠-6,催化末端单取代环氧化合物和CO2的环加成,反应24 h,产率均在80%以上;同等条件下催化末端双取代的端环氧化合物,也表现出不错的催化活性。催化1, 2-双取代环氧化合物,采用5% CaI2和5%的18-冠-6,在45 ℃和1 MPa条件下,反应48 h,可取得中高收率。18-冠-6可与钙离子形成配合物,增加了其在环氧化合物的溶解度,使I-更容易解离,增加其亲核性进而提高催化活性。该课题组进一步详细研究了无毒的聚乙二醇作助催化剂,发现碘化钙和分子量为500的聚乙二醇二甲醚(PEGDME500)组成的二元催化剂催化性能最好,而且该催化体系可有效地催化各种环氧化合物(端环氧化合物和内环氧化合物)与CO2的环加成反应。这些相转移助催化剂虽然可以增强CaI2的催化活性,可以在温和条件下催化该反应,但是反应时间较长。2018年,ZHAO等[53]发现采用含羟基的N-甲基二乙醇胺(MDEA)与CaI2在50 ℃和大气压条件下可有效地催化二氧化碳和氧化苯乙烯的环加成反应,反应6 h,产率可达98%,而且该催化剂具有较好的底物适应性和可循环使用性。MDEA不仅可以与Ca2+作用,增加其溶解度,提高CaI2的催化活性,而且自身所含羟基也能起到活化环氧化合物的作用(图9),使在温和条件具有较高的催化活性。

图9 N-甲基二乙醇胺与碘化钙催化二氧化碳和环氧化合物环加成反应机制[53]Fig.9 Proposed catalytic mechanism for the coupling of CO2 with epoxide using MDEA/CaI2 [53]
提高卤化钙催化活性的另外一种思路是采用可以活化CO2的超碱作助催化剂。HE等[54]研究了卤化钙与各种超碱组合催化CO2和环氧化合物的环加成反应,发现溴化钙和1, 8-二氮杂二环十一碳-7-烯(DBU)可以在100 ℃和大气压条件下有效地催化该反应,但是需要用到DMF做溶剂,才能表现比较好的催化活性。在5% CaI2和5% DBU作用,加入1 mL DMF,反应12 h,产率为93%。WERNER等[55]将上述两种思路整合,开发了二环己烷并-18-冠醚-6/碘化钙/三苯基膦组成的三元催化体系,该催化体系可以45 ℃和0.5 MPa下有效地催化1,2-双取代和三取代的生物基环氧化合物和二氧化碳的环加成。
相对于卤化钙,其它碱土金属卤化物用于催化该类反应较少见文献报道。2013年,REN等[56]发现MgBr2和有机碱三苯基磷(TPP)组合,可在室温无溶剂的条件下有效地催化该类反应,但需要3.5 MPa的高压和使用较多的催化剂(5% MgBr2和10% TPP)。
目前,已经开发的助催化剂虽已经可以实现碱土金属卤化物在温和的条件下催化该类反应,但同样存在催化剂用量大,反应时间长的缺点。此外,关于碱土金属卤化物催化体系的研究较少,开发集多种活性结构片段于一体的助催化剂是未来值得探究的研究方向。
3 结语
碱金属卤化物单独催化CO2和环氧化合物环加成反应的活性很低,但因其低毒、成分简单、易处理、廉价易得且稳定而成为该反应工业化的催化剂之一。目前已经开发出四类助催化剂均可以不同程度地提高其催化活性,有些甚至可以在温和的条件(大气压和接近室温条件)下,效果显著,但是还存在不少亟待解决的问题,如催化剂用量较大、反应时间长、分离催化剂实现其循环使用成本高等。碱土金属卤化物催化的研究方兴未艾,开发出的助催化剂已经可以实现温和条件的催化,但是类似于碱金属卤化物催化体系的问题同样存在。开发集多种活性基团(活化二氧化碳和环氧化合物的功能基团和配位碱金属离子和碱土金属离子的功能基团)于一体的助催化剂有望进一步提高这两类金属卤化物的催化活性,推动其在CO2资源化领域的应用。
