稳定同位素氘标记的去甲乌药碱的合成与表征
2021-08-19韩世磊陈菊玲杨立凤
韩世磊,徐 银,陈菊玲,杨立凤,张 磊
1.天津阿尔塔科技有限公司,天津 300457; 2.阿尔塔(天津)标准物质研究院有限公司,天津 300457)
去甲乌药碱最初是由Kosuge等从日本附子中分离提取,在临床上用作强心剂、利尿剂、镇痛剂和降压剂等,服用或者误服将会对运动员的成绩造成较大影响[1-2]。世界反兴奋剂机构(world anti-Doping agency, WADA)在《2019年禁用清单国际标准》中已将其明确列为β2激动剂类禁用物质。2019年8月中国体育总局反兴奋剂中心关于印发《大型赛事食源性兴奋剂》防控工作指南(暂行)的通知中,明确规定要检测沙丁胺醇、莱克多巴胺、沙美特罗、克伦丙罗、去甲乌药碱和曲托奎酚等β2激动剂。因此,有效的检测手段运用于各种食材的检测以及人体代谢物的检测尤为重要。日前,关于去甲乌药碱的检测方法主要有高效液相色谱-紫外检测(HPLC-UV)法、液相色谱-荧光检测(HPLC-FLD)法、高效液相色谱-电化学检测(HPLC-CECD)法和液相色谱-质谱(LC-MS)法等[3-6],但是其灵敏度有一定的局限性,当浓度低于最低定量限时就无法满足要求。同位素稀释质谱法(isotope dilution mass spectrometry, IDMS)采用稳定性同位素标记化合物作为内标试剂,结合了色谱的分离能力和质谱的定性能力,通过检测相应质量数的离子比值与标准比值可准确定量,同时消除样品前处理过程中引起的基质效应和回收率差异,提高检测准确度[8]。因此,稳定同位素内标试剂是该检测方法能否使用的关键因素。
当前,去甲乌药碱已经可以化学全合成,技术日趋成熟,诸多文献已对其原药的合成进行了相关报道[9-11],但关于去甲乌药碱的同位素标记化合物的文献报道较少。有文献报道了氚(T)标记去甲乌药碱化合物和去甲乌药碱-13C标记化合物的生物合成方法,但是放射性及实用性问题使其应用受到限制[12-13]。此外,有文献报道了一种关于中间体2-(3,4-二甲氧基苯)乙胺-D4的合成方法[15],其以2-(3,4-二甲氧基苯)乙腈为原料,以雷尼镍为催化剂,以D2为氘源,借助流体化学技术,合成目标产物,收率达70%,但是与氨基相邻碳上的氘代交换率仅17%,无法满足要求。同时,该方法需要使用特殊的设备以及有安全风险的氘气,因此并不适合普通实验室制备。为解决内标试剂的来源问题,本研究拟开发一条收率高、操作简便、同位素丰度高的合成路线,以期为更准确地定量检测去甲乌药碱的痕量残留提供标准内标试剂。
1 实验部分
1.1 主要仪器
IKA-RCT basic型加热型磁力搅拌器:德国IKA集团;IKA HB10型旋转蒸发仪:德国IKA集团;Bruker 600 MHz型核磁共振色谱仪:德国布鲁克公司;Agilent1260+6120型高效液相色谱-质谱联用仪:配自动进样器与DAD检测器,美国安捷伦科技公司;CHEETAH MP200型快速柱纯化仪:天津博纳艾杰尔科技有限公司;LC-IT-TOF-MS型高分辨质谱仪:日本岛津公司。
1.2 主要材料与试剂
2-(3,4-二甲氧基苯)乙腈:萨恩化学技术(上海)有限公司;重水(99.8%D)、氘代硼氢化钠(98%D)、氘代氢化铝锂(98%D):美国剑桥稳定同位素公司;4-甲氧基苯乙醛:上海毕德医药;其他试剂与药品均为市售分析纯,除特别说明外,未经处理直接使用。
1.3 实验方法
稳定同位素氘标记的去甲乌药碱(6)的合成,以2-(3,4-二甲氧基苯)乙腈为起始原料,经过碱催化的氢-氘同位素交换、氘还原、皮克特-施彭格勒(Pictet-Spengler)环化反应、脱甲基等关键反应步骤,得到四氘代的去甲乌药碱。合成路线示于图1。

图1 去甲乌药碱-D4的合成路线Fig.1 Scheme for the synthesis of higenamine-D4
1.3.12-(3,4-二甲氧基苯)乙腈-D2(2)的合成 于氮气保护下将3,4-二甲氧基苯乙腈(1)(10.0 g)溶于60 mL的干燥THF和100 mL的D2O中,加入预先干燥的K2CO3(31.0 g, 4 eq,eq表示物料当量),反应混合物于室温搅拌48 h。加入干燥的叔丁基甲基醚萃取,分液,有机相干燥、过滤,旋转蒸发浓缩后真空干燥得到2-(3,4-二甲氧基苯)乙腈-D2(2)(9.5 g),收率95.0%。1H NMR(600 MHz,Chloroform-d)δ6.87-6.82(m,2H),6.80(d,J=1.9 Hz,1H),3.88(d,J=8.5 Hz,6H)。
1.3.22-(3,4-二甲氧基)苯乙胺盐酸盐-D4(3)的合成 于氮气保护下将化合物(2)(3.0 g)溶于干燥的THF和MeOD的混合溶剂(5∶1,60 mL)中,依次加入(Boc)2O(8.0 g,2.0 eq)、氯化镍(0.23 g,1.1 eq),冰浴冷却至10 ℃左右。搅拌下分批加入氘代硼氢化钠(1.29 g,2.0 eq),然后自然升至室温,搅拌过夜。反应完毕后,加入饱和氯化铵水溶液淬灭,乙酸乙酯萃取,干燥、过滤,减压浓缩,粗品经Flash柱色谱纯化得到Boc保护的中间体,然后再加入1 mol/L的盐酸/乙酸乙酯溶液(50 mL),室温搅拌反应过夜。旋转蒸发浓缩后真空干燥得到2-(3,4-二甲氧基苯)乙胺盐酸盐-D4(3)3.8 g,收率80.0%。1H NMR(600 MHz,DMSO-d6)δ8.03(br,3H),6.93-6.83(m,2H),6.75(dd,J=8.1,2.0 Hz,1H),3.74(d,J=19.2 Hz,6H)。
1.3.36,7-二甲氧基-1-(4-甲氧基苯基)-1,2,3,4-四氢异喹啉-3,3,4,4-D4(5)的合成 于氮气保护下将化合物(3)(500 mg,1 eq)和对甲氧基苯乙醛(4)(500 mg,1.5 eq)溶于无水甲苯(20 mL)中,加入对甲苯磺酸(600 mg,1.5 eq),反应混合物加热回流3 h。反应液冷却后,用水洗涤,干燥、过滤、浓缩。粗产品经Flash柱色谱纯化得到中间体(5)400 mg,收率55.0%。1H NMR(600 MHz,Methanol-d4)δ8.50(s,1H),7.26-7.19(m,2H),6.97(d,J=8.7 Hz,1H),6.82(s,1H),6.49(s,1H),4.67(t,J=7.4 Hz,1H),3.83(d,J=11.7 Hz,6H),3.66(s,3H),3.37(dd,J=14.1,7.1 Hz,1H),3.14(dd,J=14.1,7.7 Hz,1H)。
1.3.4稳定同位素氘标记的去甲乌药碱(6)的合成 于氮气保护下将化合物(5)(200 mg)溶于无水二氯甲烷中,冷却至0 ℃。搅拌下缓慢滴加三溴化硼的二氯甲烷溶液(1 mol/L,3.8 mL,6 eq),加毕,升至室温反应3 h。反应完毕后,用氢氧化钠水溶液调节pH至12,分出水层,用盐酸调节pH至7,析出的固体经过滤、干燥,粗产物经反相色谱纯化后得到去甲乌药碱-D4,89.2 mg,白色固体,收率51.4%,HPLC纯度98.0%,氘丰度为96.5%。HRMSm/z:[M+H]+=276.152 8(理论值276.153)。1H NMR(600 MHz,Methanol-d4)δ7.14(d,J=8.5 Hz,1H),6.81(d,J=8.5 Hz,1H),6.63(s,1H),6.62(s,1H),4.58(dd,J= 8.9,5.6 Hz,1H),3.37(dd,J=14.6,5.6 Hz,1H),2.97(dd,J=14.6,9.0 Hz,1H)。
2 结果与讨论
2.1 去甲乌药碱-D4(6)的合成
2.1.12-(3,4-二甲氧基苯)乙腈-D2(2)的合成 在化合物(2)的合成研究中,对不同的氘源及碱作为氢-氘交换的反应条件进行优化,结果列于表1。从表1中可以看到,无论是有机金属碱还是无机碱,底物交换反应后的同位素丰度均较高,但是有机金属试剂作为碱时,反应体系有较多的杂质,收率相对较低,而用氢氧化钠或者氘氧化钠作碱时,产物收率明显降低,推测可能是由于底物分子中的氰基水解所致,综合实验操作的简便性及原辅材料廉价易得性,最终选择K2CO3/THF/D2O体系进行该步氘-氢交换反应。
此外,针对表1中反应条件3的碱和重水的使用量、反应温度及反应时间对反应收率和同位素丰度的影响进行研究,结果列于表2。由表2结果可知,反应温度对同位素丰度影响较小,但是在较高温度下反应收率有降低的趋势,而增加重水的使用量和反应时间有利于提高产物的同位素丰度。
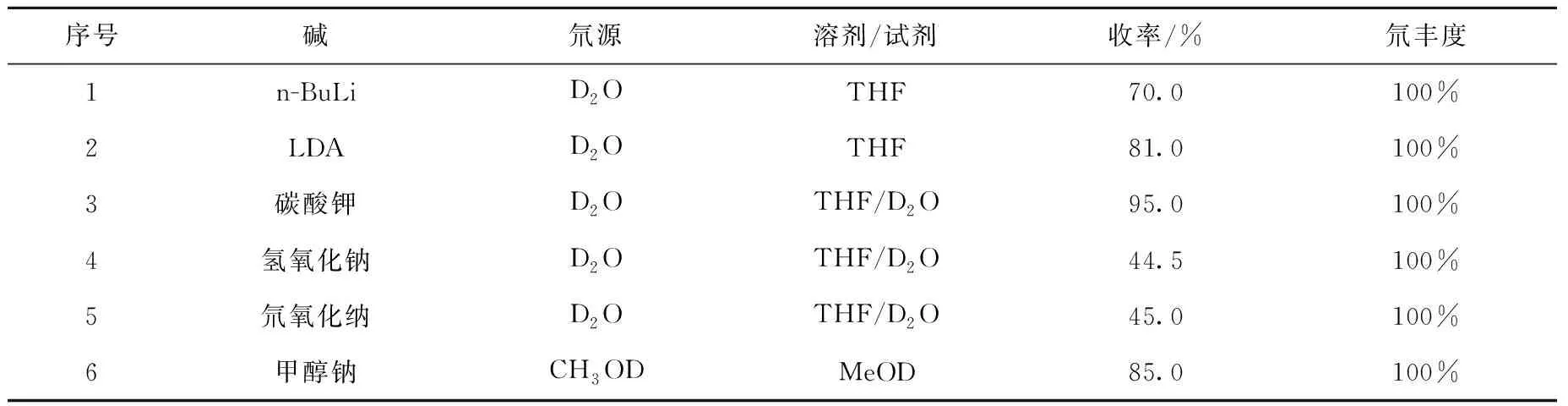
表1 化合物(2)合成的反应条件筛选Table 1 Reaction condition screening for the synthesis of compound (2)
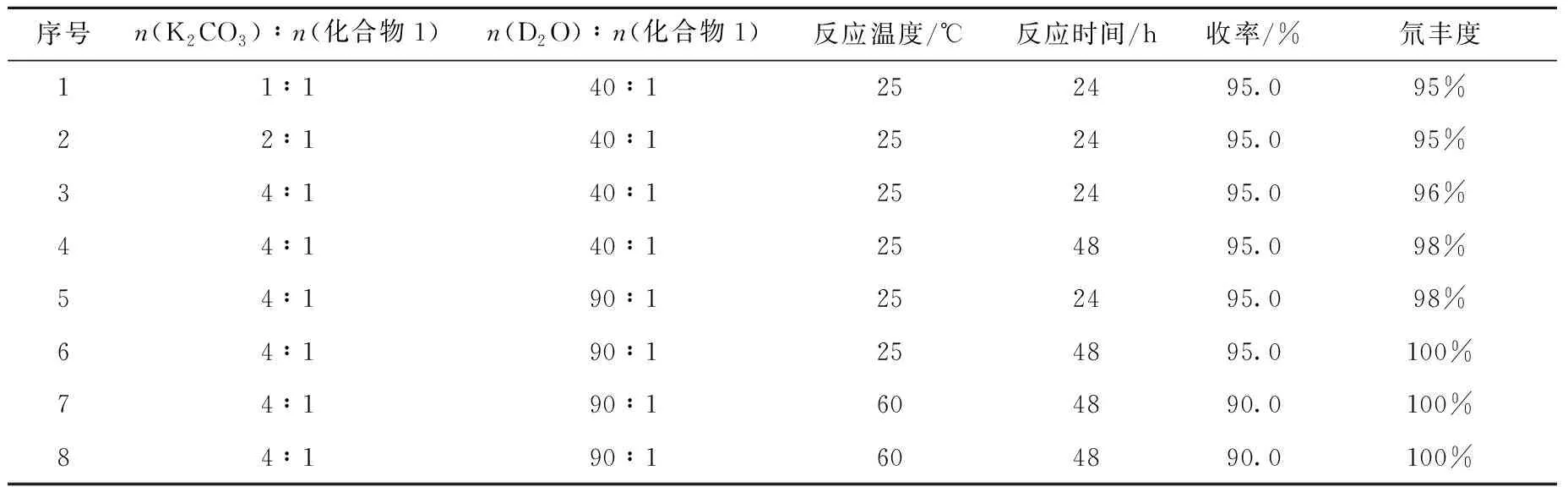
表2 试剂与底物的物质的量比对化合物(2)合成的影响Table 2 Effect of materials ratio on for the synthesis of compound (2)
2.1.22-(3,4-二甲氧基)苯乙胺盐酸盐-D4(3)的合成 在化合物(3)的合成研究中,针对不同还原剂、溶剂对反应的影响进行研究,结果列于表3。使用四氢铝锂作还原剂时,由于淬灭过程中产生的铝盐对产物的包裹,导致收率较低;而硼氢化钠作为还原剂并进行“一锅法”投料,使还原生成的胺原位生成Boc保护的胺,然后脱去Boc保护基得到目标化合物(3),可极大地提高产物收率。此外,使用四氢呋喃-甲醇做共溶剂可极大地提高反应速率,而用氘代甲醇(CH3OD)替换甲醇(CH3OH),可有效避免氢-氘交换,将产物的同位素丰度提高约97%。

表3 不同还原剂对化合物(3)合成的影响Table 3 Effect of reducing agents on the synthesis of compound (3)
2.1.36,7-二甲氧基-1-(4-甲氧基苯基)-1,2,3,4-四氢异喹啉-3,3,4,4-D4(5)的合成 化合物(5)的合成属于Pictet-Spengler环化反应,本文对不同的酸性试剂及溶剂对反应的影响进行研究,结果列于表4。对甲苯磺酸作为质子酸体系催化效果较好,而其他酸性试剂如盐酸体系结果不理想。
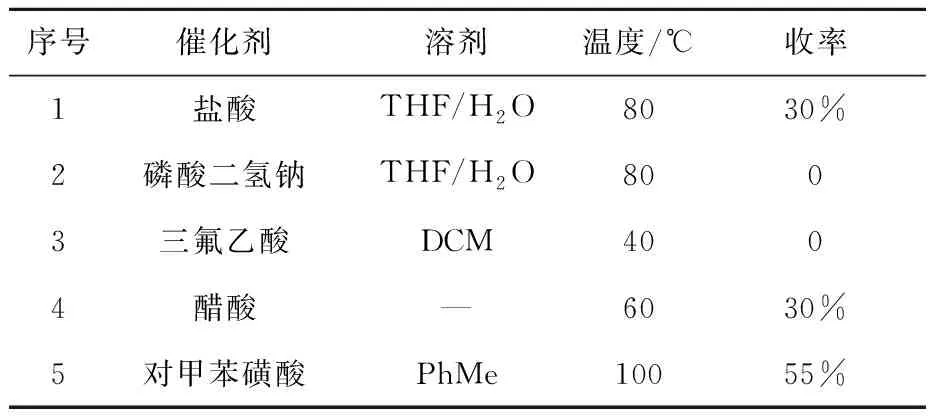
表4 催化剂对化合物(5)合成的影响Table 4 Effect of catalyst on the synthesis of compound (5)
2.1.4去甲乌药碱-D4(6)的合成 使用氢溴酸作为脱甲基化试剂,产物色素较重,对产品的质控不利,而使用三溴化硼作为脱甲基化试剂有效避免了该问题。此外,在后处理过程中发现,因该化合物分子同时含有酸性基团(酚羟基)和碱性基团(氨基),体系的pH对结晶析出及收率至关重要,而且化合物含极性基团较多,其在水中具有一定的溶解性,水的使用量对其收率也有较大影响。
2.2 去甲乌药碱-D4(6)的鉴定与表征
2.2.11H NMR 本文中所有化合物均通过1H NMR和HRMS进行鉴定,检测结果与结构相符。去甲乌药碱-D4(6)的1HNMR谱图示于图2。δ7.14 ppm,6.81 ppm两个双重峰为4-羟基苯甲基片段中苯环上的氢,6.63 ppm、6.62 ppm两个单峰为1,2,3,4-四氢异喹啉结构中苯环上两个氢,δ4.58 ppm峰为1,2,3,4-四氢异喹啉结构中1-位手性次甲基氢与相邻的亚甲基耦合的核磁信号。与该手性中心相连的4-羟基苯甲基片段中苄位亚甲基上的两个质子(图3)为化学位移不等价,在δ3.37 ppm、2.97 ppm表现为两个多重峰,与文献报道一致[15]。去甲乌药碱-D4(a)与非标记的去甲乌药碱(b)的氢核磁谱图对比示于图4,由于四氢异喹啉3-位和4-位亚甲基上的质子被氘原子取代,因此在去甲乌药碱-D4(6)的氢核磁谱图上无信号峰。
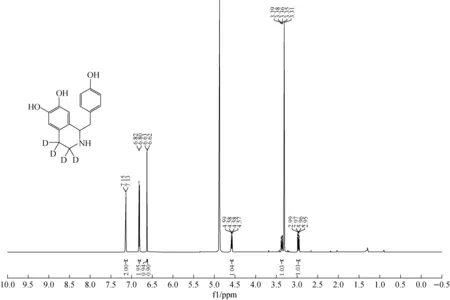
图2 去甲乌药碱-D4的1H NMR谱图Fig.2 1H NMR spectra of higenamine-D4

图3 模拟的去甲乌药碱的立体结构Fig.3 Simulated 3D structure of higenamine

图4 1H NMR核磁谱图比对Fig.4 1H NMR comparison spectra of higenamine-D4 and higenamine
2.2.2HRMS确证及同位素丰度确认 去甲乌药碱-D4的精确相对分子质量通过飞行时间质谱得到确认。通过质谱的结果(图5)可知,目标化合物的分子离子峰[M+H]+=276.152 8,与理论计算结果[M+H]+=276.153 8相符。

图5 去甲乌药碱-D4的HRMS谱图Fig.5 HRMS spectrum of higenamine-D4
同时,产物经串联质谱分析,用“质量簇分类法”[16-17]计算,去甲乌药碱-D4氘同位素的丰度为96.5%,具体计算过程如下。
去甲乌药碱-D4的质谱峰簇为:m/z=272~278,非氘标记的化合物天然丰度分布的占比(归一化后)应为A272∶A273∶A274=0.84∶0.15∶0.01。将所采集到的质谱图中m/z=272、273、274、275、276的峰强度数据归一化后分别记为A0、A1、A2、A3、A4,代入方程组(1)计算,解xj(j=0、1、2、3、4)。
A0=0.84x0=0
A1=0.15x0+0.84x1=0
A2=0.01x0+0.15x1+0.84x2=0
(1)
A3=0.01x1+0.15x2+0.84x3=11.4
A4=0.01x2+0.15x3+0.84x4=72.6
从上述方程组中求得x0=0,x1=0,x2=0,x3=13.57,x4=84.01。
去甲乌药碱-D4的同位素丰度值以E表示,按公式(2)计算得到同位素丰度,即氘原子标记率E=96.5%。
(2)
式中:xj表示标记j个D原子的去甲乌药碱-Dj分子的摩尔分数,其中j=0、1、2、3、4。
3 结论
本文以2-(3,4-二甲氧基苯)乙腈为起始原料,以廉价易得的重水为稳定同位素标记源,经过氢-氘交换,再经氘代金属还原剂还原得到关键中间体2-(3,4-二甲氧基苯)乙胺盐酸盐-D4,再经关环、脱保护基等步骤,合成了去甲乌药碱-D4。其中关键是氢-氘交换和氘代还原两个过程对化合物同位素丰度的控制,经过条件优化,最终合成得到的关键中间体的同位素丰度达到了预期,且后续反应并未有明显的同位素丰度稀释现象。本文所合成的目标化合物能作为内标试剂满足去甲乌药碱的定性与定量分析。
