气相色谱-质谱法测定黄瓜中毒死蜱残留量的测量不确定度评定
2021-08-11汪春明施鹏斐
汪春明,张 洋,施鹏斐,杨 波
(1.中检科(北京)测试技术有限公司,北京 100123;2.中国检验检疫科学研究院综合检测中心,北京 100123)
测量不确定度是根据所用到的信息,表征赋予被测量值分散性的非负参数[1],是与测量结果相联系的参数[2]。简单而言,不确定度是指由于测量过程中误差的存在,对被测量值不能肯定的程度,代表测定值的可信区间,是衡量实验室检测能力的重要指标,通过对检测不确定度的主要影响因素进行评价,以期为测量数据的准确性提供科学依据,确保检验结果的可靠性[3−7]。
不确定度从Eisenhart提出,到1993年ISO成文刊出,到2011年CNAS明确规定“检测实验室应有能力对每一项有数值要求的测量结果进行测量不确定度评估”,再到CNAS-GL006的进一步细化完善[8−11]。农药残留分析实验室开始对测定值的不确定度重视起来。然而,有关农药残留测量不确定度方面的研究相对较少,且对影响测量值不确定度的各分量认识不统一[12−18]。造成这种原因,一方面是由于各检测实验室人员技能、前处理方法、仪器、耗材等因素的差异;另一方面是由于实验室未具备合理评估测量不确定度的能力。
为系统科学地评估测量不确定度,本研究依据GB 23200.113-2018《食品安全国家标准 植物源性食品中208种农药及其代谢物残留量的测定》[19],结合自身实验室的具体操作步骤,对黄瓜中毒死蜱农药残留量的测定结果进行不确定度评定,以期望为农药残留检测实验室进行不确定度评定时提供参考。
1 材料与方法
1.1 材料与仪器
黄瓜 来源于市售样品;毒死蜱标准品(纯度99.64%) 德国DrEhrenstorfer公司;乙腈、乙酸乙酯 色谱纯,德国Meker公司;氯化钠、无水硫酸镁、柠檬酸钠、柠檬酸氢二钠 分析纯,国药集团化学试剂有限公司;PSA Agilent科技有限公司。
GCMS-TQ8040 日 本Shimadzu公 司;HP-5MS UI气相色谱柱(30 m × 0.25 mm,0.25 μm)美国Agilent公司;移液枪 德国Eppendorf公司;SR-2DS振荡器 日本TAITEC公司;CR21N离心机日本Hitachi公司;VORTEX GENIE 2涡旋混匀器美 国Scientific Industries公 司;XS105 &PL303分析天平 瑞士Mettler Toledo公司;EVA50A氮吹仪 北京普立泰科仪器有限公司。
1.2 实验方法
1.2.1 标准溶液配制 标准储备液的配制:准确称取毒死蜱15.05 mg于10 mL容量瓶中,由丙酮定容至刻度,得到1505 μg/mL的标准储备液。
标准中间液的配制:移取毒死蜱66.4 μL标准储备液于10 mL容量瓶中由乙酸乙酯定容至刻度,得到10 μg/mL毒死蜱标准中间液。
工作曲线的绘制:吸取10 μg/mL毒死蜱标准中间液80、120、160、200、240、280 μL于10 mL容量瓶,由乙酸乙酯定容至刻度;将1 mL空白基质溶液氮气吹干,分别加入1 mL上述工作溶液复溶,即得到0.08、0.12、0.16、0.20、0.24、0.28 μg/mL标准工作溶液。
1.2.2 样品添加浓度 根据GB 2763-2019[20]黄瓜中毒死蜱最大残留限量为0.1 mg/kg,本实验添加浓度亦为0.1 mg/kg。
1.2.3 样品前处理方法 参照GB 23200.113-2018《食品安全国家标准植物源性食品中208种农药及其代谢物残留量的测定气相色谱-质谱联用法》[19]进行样品前处理,供试黄瓜全瓜(去柄)匀浆,待测。
准确称取10 g样品(精确至0.01 g)于50 mL离心管中,加入10 mL乙腈、1 g氯化钠、4 g无水硫酸镁、1 g柠檬酸钠、0.5 g柠檬酸氢二钠及1颗陶瓷均质子,振荡提取30 min后,10000 r/min离心3 min。吸取6 mL上清液加入内含150 mg PSA和900 mg无水硫酸镁的10 mL离心管中,涡旋混匀1 min,5000 r/min离心5 min,准确吸取2 mL上清液于10 mL螺口接收管中,40 ℃水浴中氮气吹至近干,加入1 mL乙酸乙酯复溶,过有机相膜,GC/MS/MS测定,外标法定量。
1.2.4 仪器检测条件 色谱柱温度:初始温度40 ℃保持1 min,40 ℃/min升至120 ℃,再以5 ℃升温至240 ℃,再以12 ℃/min升温至300 ℃,保持6 min;进样口温度280 ℃;进样量1 μL,载气为氦气,流速1.0 mL/min;不分流进样;EI电子轰击源,能量70 eV;传输线温度280 ℃,溶剂延迟3 min。毒死蜱定量离子对196.9>169.0,碰撞能量15 eV,定性离子对198.9>171.0,碰撞能量15 eV。
1.3 数学模型
毒死蜱残留量的质量分数ω结果计算公式:

式中,ω:样品中毒死蜱的质量分数,mg/kg;c:由校准曲线计算的样品溶液的浓度,μg/mL;V:样品定容体积,mL;V1:提取溶液的体积,mL;V2:分取溶液的体积,mL;m:称取样品的质量,g。
样品中毒死蜱质量分数的测定值除受上述计算公式中c、V、V1、V2、m因子的影响外,还与样品的均匀性和前处理过程的一致性相关,考虑其影响因素,公式中需加入修正因子frep,则上式农药的质量分数ω与输入量的函数关系(数学模型)见下式:

由图1可知,样品中毒死蜱残留量合成标准不确定度[urel(ω)]见式:
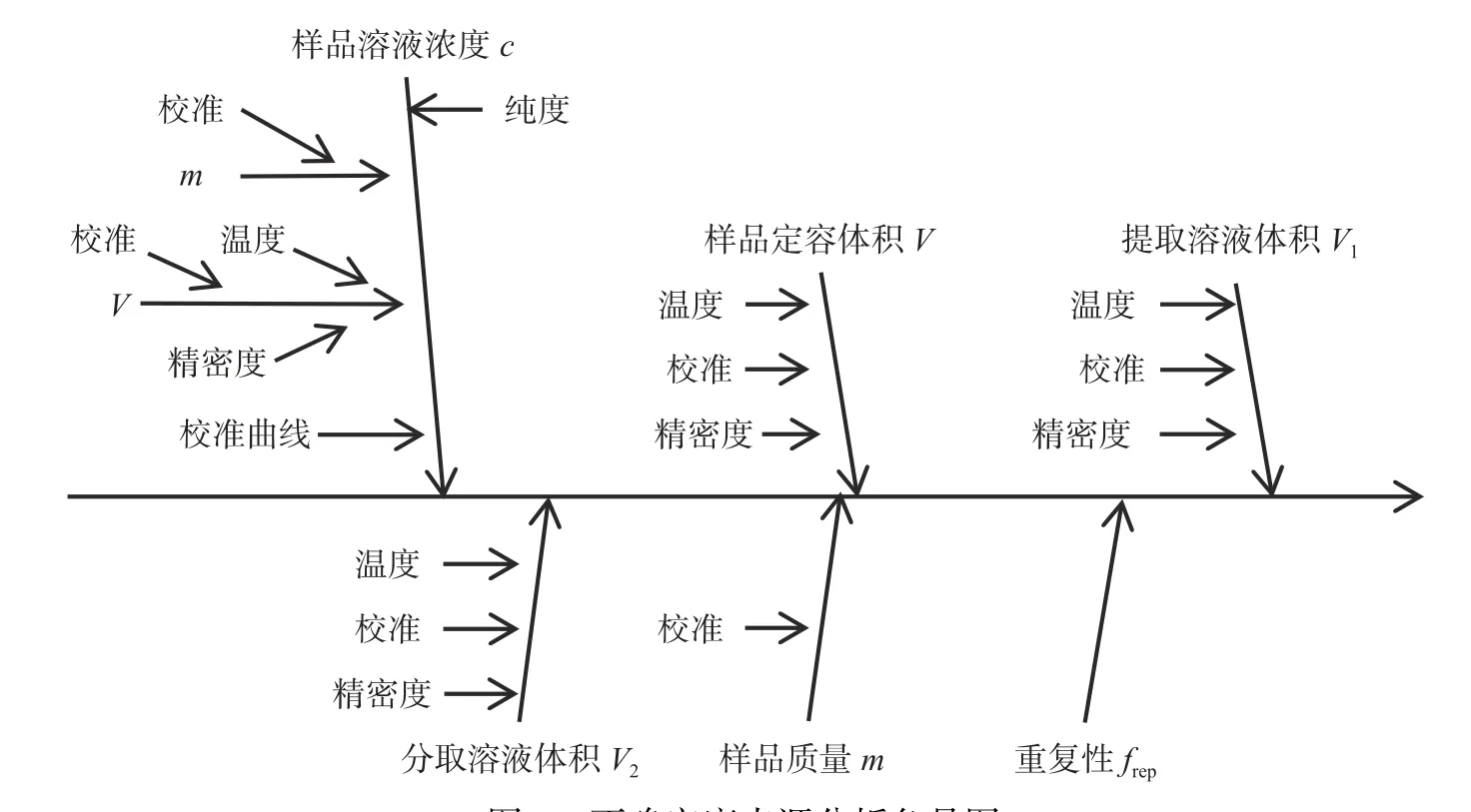
图1 不确定度来源分析鱼骨图Fig.1 Analysis fishbone diagram of the uncertainty source

1.4 数据处理
数据的收集与计算在Excel中进行,相关公式的编辑由MathType数学公式编辑器6.9b(简体中文版)生成,鱼骨图在Word中手动编辑形成。
2 结果与分析
2.1 识别和分析不确定度来源
不确定度评定方法大致分为两类:英国皇家化学会分析方法委员会提出的自上而下(Top-down)和以ISO/IEC Guide98-3: 2008《测量不确定度表述指南》为代表的自下而上(Bottom-up)两类。其中自上而下(Top-down)评定方法有精密度法、控制图法、线性拟合法和经验模型法四种,是对很长周期内的实验室间协作定值/实验室内部质量控制、方法验证、能力验证和已经发表文献的精确数据进行不确定度评定,该方法需要从整体上、通过数月、数年等一段时间积累的大量数据支持来直接进行测量不确定性评估;Bottom-up方法有GUM和MCM两种,它基于检测过程并对检测过程中每一个输入量进行评估,之后建立数学模型,对测量不确定度的来源进行识别和量化,从而计算合成不确定度,是目前较通用的一种方法[21−24]。因此本研究基于Bottom-up方法进行不确定度评定。结合实验过程和数学模型分析。样品农药含量测定结果的不确定度主要来源于:称样、样品前处理、样品溶液的浓度计算、样品本身均匀性及前处理差异4个方面所产生的不确定度。
2.2 不确定度分量的评定
2.2.1 样品溶液中毒死蜱浓度的不确定度
2.2.1.1 配制标准溶液引入的不确定度 配制标准溶液引入的不确定度主要来源于标准物质、标准物质称量、标准物质定容。
标准物质:依据标准物质证书所列信息,毒死蜱的不确定度为0.70%,置信水平P=95%,包含因子k=2,一般认为标准物质证书给出的为扩展不确定度,所以毒死蜱标准物质的相对标准不确定度[urel(s)]为:

标准物质称量:用电子天平称取毒死蜱标准物质15.05 mg,天平校准本身有两个可能的不确定度来源:灵敏度和校准函数的线性。如果称量是用同一台天平且称量范围很小,则灵敏度带来的不确定度可忽略不计。天平制造商自身的不确定度建议采用矩形分布将线性分量转化为标准不确定度[11]。根据检定证书得知天平的示值误差为0.1 mg,所以标准物质称量引入的相对标准不确定度[urel(ms)]为:
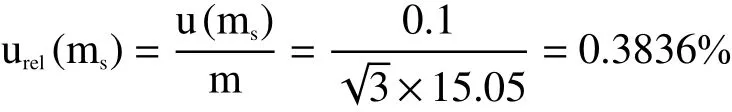
标准储备液配制定容:使用玻璃量器引入的相对不确定度[urel(Vr)],标准溶液配制时用到了10 mL容量瓶,查JJG 196-2006[25]10 mL(准确度等级为A)容量瓶容量允差±0.02 mL,由于其没有给定置信水平,且认为极端值可能时,通常假定其为矩形分布,10 mL容量瓶读数误差引入的相对标准不确定度[urel(Vrk)]为:

由于容量瓶(硼硅玻璃体积膨胀系数1.0×10−5℃−1)随温度变化的范围远小于定容溶剂随温度变化而引起的体积变化,故只考虑定容溶剂体积膨胀系数而引入的不确定度,固体标准品由丙酮溶解并定容至10 mL容量瓶后,分取一部分后由乙酸乙酯溶解并定容至10 mL,实验室温度变化范围(20±4)℃,丙酮在(20±4)℃条件下的体积膨胀系数0.143% ℃−1,乙酸乙酯在(20±4)℃条件下的体积膨胀系数为0.138% ℃−1,服从矩形分布,则温度对丙酮和乙酸乙酯体积变化的相对标准不确定度[urel(Vrw丙酮)、urel(Vrw乙酸乙酯)] 分别为:
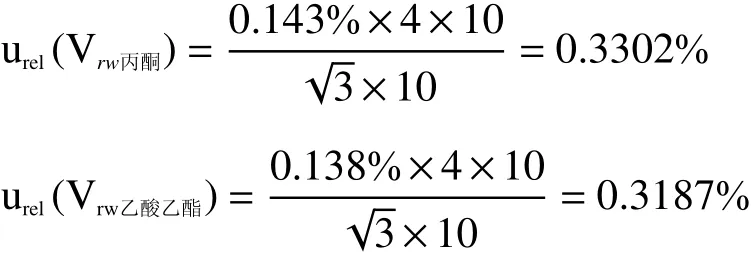
容量瓶在读数的时候也会引入误差,对典型的10 mL容量瓶反复充满10次并进行称量,得到的实验标准偏差为0.01 mL,则标准不确定度分量[u(Vrd)]为:

10 mL容量瓶读数误差(重复性误差)引入的相对标准不确定度[urel(Vrd)]为:

则由丙酮和乙酸乙酯定容至10 mL时,容量瓶引入的不确定度[urel(Vr丙酮)、urel(Vr乙酸乙酯)]如表1所示。

表1 标准系列溶液配制中玻璃量器引入的不确定度Table 1 Relative standard uncertainty from working glass contain in standard solution preparation
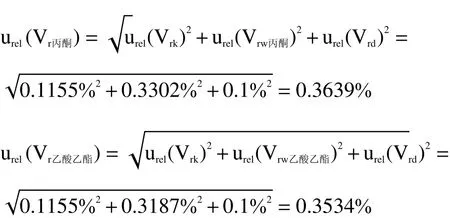
100 μL移液器:由检定证书得知,其容量误差为2%,则由定值误差引入的相对不确定度为:

丙酮在(20±4)℃条件下的体积膨胀系数为0.143% ℃−1,乙酸乙酯在(20±4)℃条件下的体积膨胀系数为0.138% ℃−1,则由温度变化引入的相对标准不确定度为:

由检定证书得知100 μL移液器的测量重复性1.5%,则由读数引入的相对标准不确定度为:


同样的,其余移液器引入的相对标准不确定度详见表2。
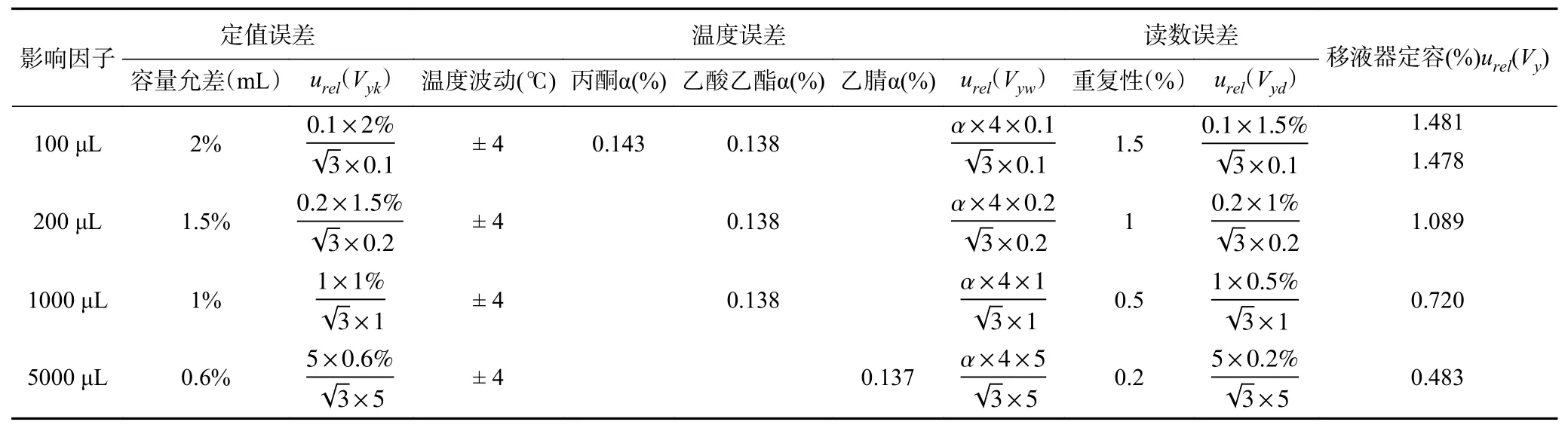
表2 标准系列溶液配制中移液器引入的不确定度Table 2 Relative standard uncertainty from locomotive pipette in standard solution preparation
系列标准溶液配制过程中一共用到10 mL容量瓶8次(其中1次为丙酮定容,7次为乙酸乙酯定容),100 μL移液器2次(一次为乙酸乙酯、一次为丙酮),200 μL移液器7次,1000 μL移液器6次,则系列标准溶液配制过程中引入的相对标准不确定度为:
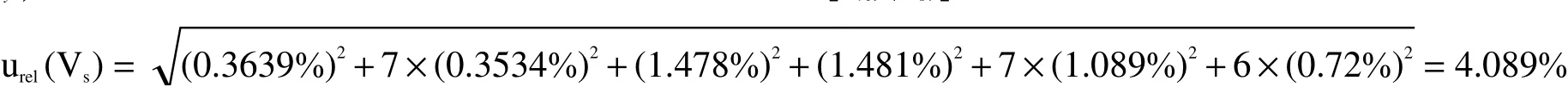
2.2.1.2 校准曲线拟合的不确定度分量 被测物质在仪器上的信号强度与其对应的浓度成正比,通过测定已知浓度溶液的信号强度,利用最小二乘法将响应值和浓度对应的线性关系拟合成一条直线,再由未知样品的响应值推算对应的浓度,然而并非所有参与校准曲线的浓度点都会落到标准曲线之上,因此校准曲线也会引入不确定度。本研究由空白基质溶液配制六个点的校准曲线溶液,浓度范围分别为0.08、0.12、0.16、0.20、0.24、0.28 μg/mL,每个点平行测定3次,统计数据见表3。
通过最小二乘法将响应值与浓度的对应关系拟合成的工作曲线回归方程为:A = bc+a,
式中:A:测量溶液的色谱峰面积;a:工作曲线的截距;b:工作曲线的斜率;c:测量溶液中农药的浓度。由校准曲线浓度及相应面积拟合的回归方程为:

当未知样品根据响应值带入校准曲线得到浓度值的过程中也会引入不确定度,则由工作曲线变动性引起的标准不确定度分量[u(c0)]为:
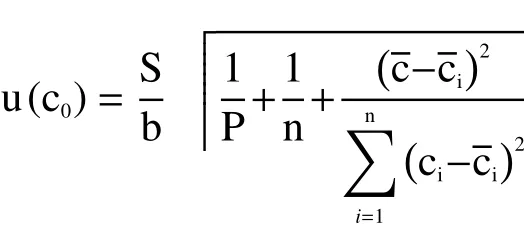
S:标准溶液峰面积的残差标准偏差;b :校准曲线的斜率;P:被测样品平行测定次数(10次);n:参与校准曲线拟合的点数(6水平×3平行=18);被测样品平行测定质量浓度的平均值;校准曲线溶液质量浓度的平均值μg/mL;ci:系列标准曲线中各溶液浓度的实测值。详细数据见表3,其中S的计算公式如下:


表3 标准曲线引入的不确定度Table 3 The uncertainty resulting from the fitting process of standard curve
2.2.2 样品质量的不确定度 称量样品所使用的天平为0.001 g精度的分析天平,称取的样品质量为10.047 g(10次称量的平均值,天平检定记录所示其最大允许误差为±0.005 g,按矩形分布则称样量引入的相对标准不确定度

2.2.3 样品前处理引入的不确定度 提取:样品提取液加入步骤为用5 mL移液器分两次(5 mL×2)准确加入10 mL乙腈。根据JJG 646-2006《移液器检定规程》的规定,移液器的各种不确定度分量都被认为服从均匀分布。5 mL移液器最大容量允差为0.6%,则由定值误差引入的相对标准不确定分量为移液器重复性偏差0.2%,则由读数误差带入的相对标准不确定度为实验室温度变化范围(20±4)℃,乙腈在(20±4)℃条件下的体积膨胀系数为0.137% ℃-1,则由温度变化引入的相对标准不确定度分量为移液器引入的相对标准不确定度为详见表2。故提取液移取引起的相对标准不确定度为
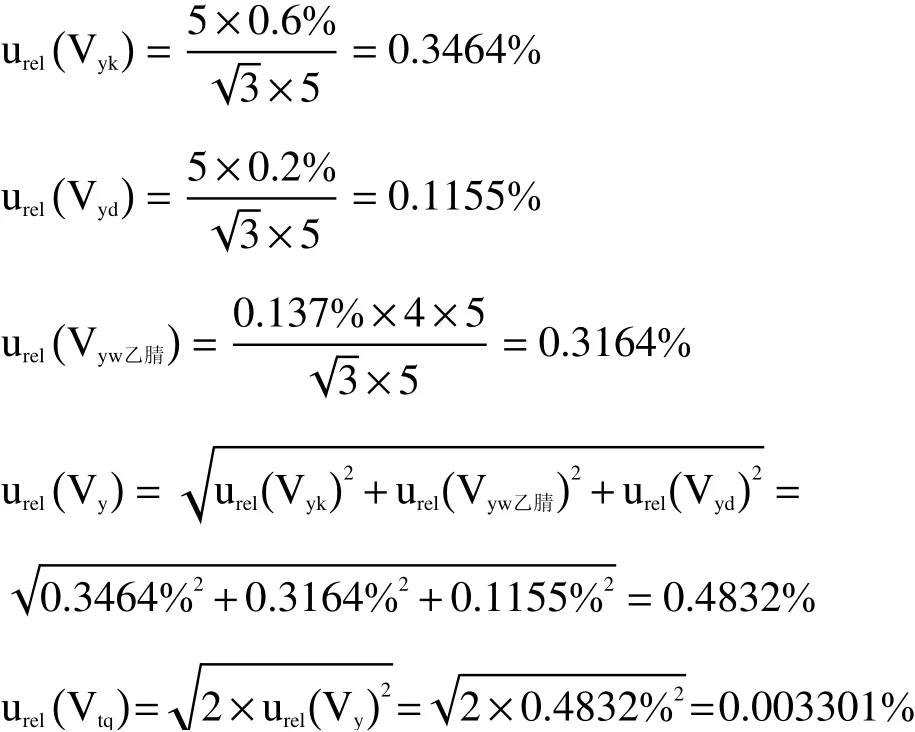
分取:提取液分取步骤为用5 mL移液器分两次(3 mL×2)共分取6 mL乙腈提取液净化,之后再用5 mL移液器移取2 mL氮吹;其它不确定度分量影响变化与样品提取一样,则由提取液分取引入的不确定度也为
定容:浓缩定容是将分取液氮吹至近干;然后用1.0 mL移液器移取1.0 mL乙酸乙酯复溶浓缩液。1.0 mL移液器最大允差为1%,则由定值误差引入的相对标准不确定度为1.0 mL移液器重复性偏差为0.5%,则由读数误差带入的相对标准不确定度为乙酸乙酯在(20±4)℃条件下的体积膨胀系数为0.138% ℃−1。则由温度变化引入的相对不确定度分量为0.3187%。故提取液移取引起的相对标准不确定度为urel(Vdr)=0.720%。具体由1.0 mL移液器引入的相对标准不确定度计算详见表2。
2.2.4 测量重复性引入的不确定度 测量重复性是对整个实验系统的不确定性进行评估,包括样品本身均匀性、人员操作熟练程度、环境温度、仪器稳定性及前处理方法差异等引起的不确定度。从总体效果看,这项不确定度分量实际上包含了多项可以用B类方法评定的不确定度分量,然而,如果运用单一的B类评定方式,就需要对样品、人员、环境、仪器、方法等影响因素分别进行评定,通过分析各个误差的来源并对各分量的不确定度进行量值,之后再通过一定的关系将所有单个因素进行合成,得到其合成不确定度,然而由于人员、环境等因素难于量化且计算难度大,运用B类方法对其进行不确定度评定变得极为不可能,如果将测量重复性不确定度采用A类评定,采用重复性测量的标准方差计算,则简化了测量结果不确定度的评定步骤[27−28],采用A类评定的数据不能低于7个,为了评定不确定度的数据准确可靠,研究者取同一阴性样品,按0.1 mg/kg的水平进行加标回收试验,测定了10个平行加标样品,测定结果见表4。

表4 测量重复性引入的不确定度Table 4 The uncertainty resulting from the repeatability
2.3 合成标准不确定度的计算
对上述各不确定度分量进行总结,结果见表5,将相对标准不确定度各分量通过1.3节样品中毒死蜱残留测定结果的合成标准不确定度公式进行合成,即得到测量农药的合成标准不确定度。
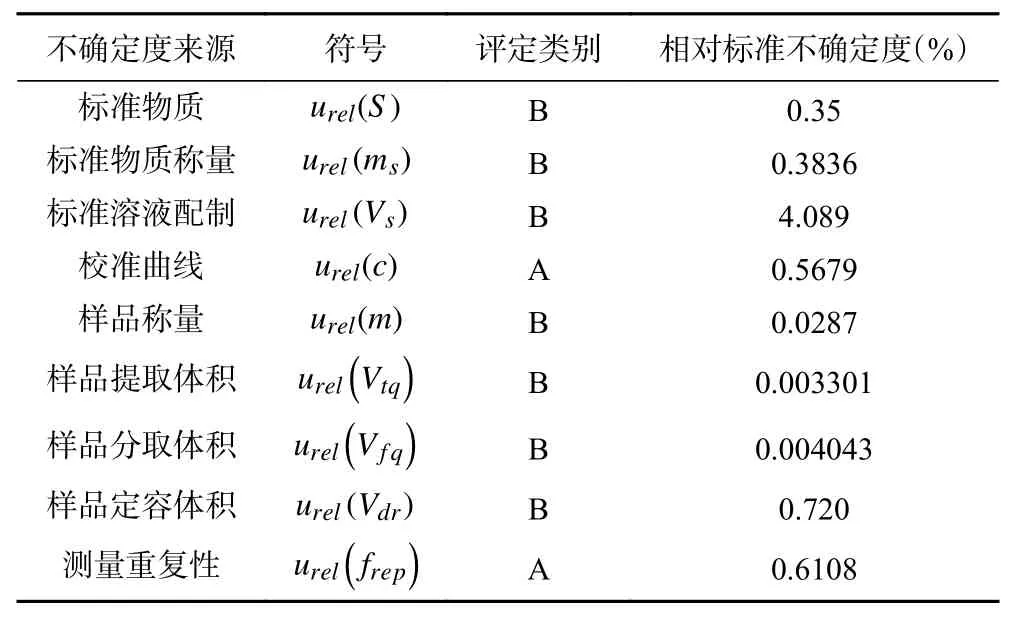
表5 合成标准不确定度的构成Table 5 The statistics of the relative standard uncertainty
不确定度传播公式:
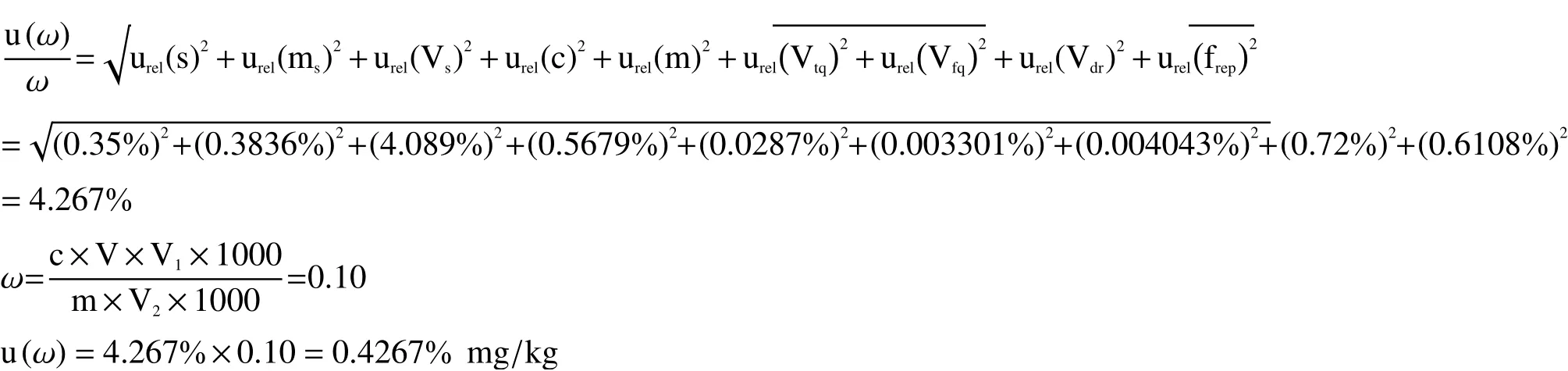
2.4 测量结果的扩展不确定度
应测量不确定度指南的要求,一般检测实验室在做不确定度评定的时候,普遍取95%的置信水平,包含因子k = 2,则扩展不确定度

3 讨论与结论
毒死蜱是一种有机磷类非内吸性广谱杀虫、杀螨剂;具有良好的触杀、胃毒和熏蒸作用,可防治多种害虫;而且可与多种杀虫剂混用以达到增效的作用,相对而言毒性低。正是因为其本身的这种特性而被广泛应用于农业生产中。本文在研究日常果蔬的检测过程中发现,毒死蜱易检出、且易超出GB 2763[20]《食品安全国家标准食品中农药最大残留限量》。当毒死蜱检出、且位于限量值附近时,如何才能更好的对受检样品进行准确判定,便成了检测工作者首要面对的难题。本研究依据食品安全国家标准GB 23200.113-2018[19],结合自身实验室的具体操作步骤,参考CNAS-GL 006、计量检定(技术)规范等相关标准,对黄瓜中的毒死蜱农药残留量进行不确定度评定。结果显示黄瓜中毒死蜱的检测结果表示为ω=(0.10±0.01)mg/kg,k = 2。在本研究所有不确定度引入的分量中,标准工作溶液配制引入的不确定度分量最大,可见在实验室内部质量控制中,标准工作液的配制过程宜充分重视。通过使用容量误差更小的容量瓶、降低实验操作员的人为读数误差、适当减少移液器使用数量、缩小实验环境温差波动范围,可适当降低实验结果的不确定度。
化学分析中对不确定度的需求日益增强,尤其是测定值在限量值附近时。本不确定度评定给定的思路和方法也适用于其他利用气相色谱质谱联用仪测定农残时的不确定度评定。建议有能力的实验室可利用标准物质或其他性质稳定的样品,按照规定的方法步骤,在不低于10次的独立测量条件下,按上述程序评定待测物的不确定度。
