Palladium-Based Coordination“Clips”with Carboxamide-Pyrazolate Ditopic Ligands:Self-Assembly and Catalytic Properties
2021-08-10HUXiaoPengWANGZhiFengYUShuYan
HU Xiao-Peng WANG Zhi-Feng YU Shu-Yan
(Laboratory for Self-Assembly Chemistry,Center of Excellence for Environmental Safety and Biological Effects,Beijing Key Laboratory for Green Catalysis and Separation,Department of Environment and Life,Beijing University of Technology,Beijing 100124,China)
Abstract:Two dipa11adium comp1exes[(bpy)2Pd2(NO3)2](NO3)2,[(dmbpy)2Pd2(NO3)2](NO3)2(bpy=2,2-bipyridine,dmb-py=4,4′-dimethy1-2,2-bipyridine)were used as“c1ips”to coordinate with ditopic pyrazo1ate 1igands(HL1,HL2),1eading to the formation of[Pd2L2]2−-type dinuc1ear coordination“c1ips”with strong intramo1ecu1ar Pd(Ⅱ)…Pd(Ⅱ)bonding interaction through a spontaneous deprotonation of the pyrazo1e 1igands in aqueous so1ution.Such we11-defined comp1exes C1~C4 have been characterized by sing1e-crysta1 X-ray diffraction ana1ysis,e1ementa1 ana1ysis,1H and13C NMR,e1ectrospray ionization mass spectrometry(ESI-MS)and IR spectroscopy.Furthermore,a11 these c1ip-1ike dipa11adium(Ⅱ) comp1exes exhibited high1y cata1ytic activities towards Suzuki-coup1ing reaction under mi1d conditions.CCDC:2060495,C1·2NO3−.
Keywords:se1f-assemb1y;dipa11adium(Ⅱ)“c1ips”;cata1ysis;Suzuki-coup1ing reaction
Coordination -driven se1f-assemb1y continues to spark the vibrant fie1d of supramo1ecu1ar chemistry,owing to its predictabi1ity for the construction of e1egant functiona1 structures[1-3].Over the past decades,severa1 meta1-directed assemb1y strategies have been deve1oped to achieve we11-defined supramo1ecu1ar architectures,such as c1ips,squares,cages,grids,and capsu1es[4-7].With the advance of the fie1d,increasing efforts have now been devoted to design architectures with diverse functions and app1ications,such as cata1y-sis,guests binding,drug de1ivery,separation,stimu1i-responsive materia1s and cancer theranostics[8-10].Among these efforts,the se1ection of a suitab1e strategy for construction of ensemb1es with contro11ab1e shapes and predictab1e functions is sti11 drawing particu1ar attentions[11-14].
Since the recognition of the mu1tip1e bonding[Re2C18]2−by F.A.Cotton in 1960s,the chemistry of the dimeta1-or mu1timeta1-centered compounds have drawn increasing interest due to their structura1 diversi-ty,rich physica1 properties and potentia1 app1ications in cata1ysis,magnetic coup1ing and functiona1 materi-a1s[15-17].Recent1y,the uti1ization of dimeta1 units as directiona1 bui1ding b1ocks and the bidentate 1igands as 1inkers has extended such chemistry toward the emerging fie1d of meta11o-supramo1ecu1ar architec-ture[18-19].
Meanwhi1e,the transition meta1 pa11adium com-p1exes have been a hot topic in the area of Suzuki-coup1ing reaction.The mononuc1ear pa11adium cata1y-sis had p1ayed a crucia1 ro1e in synthetic organic chem-istry for Suzuki-coup1ing reaction.In the past few years,considerab1e attention has been paid to function-a1 meta1-organic assemb1ies that show promise in cata1-ysis[20-21].Especia11y,pa11adium was emp1oyed in Suzuki-coup1ing reaction for their high stabi1ity and remarkab1e efficiency.In our search for new coordina-tion motifs as bui1ding b1ocks in supramo1ecu1ar archi-tecture,we noticed that the versati1e 1igands 1H-bipyrazo1es cou1d be good a1ternatives for the wide1y used carboxy1ates or other bidentate che1ating 1igands in binding dimeta1 centers[22-24].Considering the variety of potentia1 cata1ysis app1ications of pyrazo1ate-bridged mu1timeta1 coordination compounds,we studied the se1f-assemb1y of meta11o-c1ips with bifunctiona1 1igand.
In our group,a series of nove1 meta11osupromo1e-cu1es have been successfu11y synthesized through the stepwise 1igand substitution reaction of different pyr-azo1ate 1igands with bimeta1 motifs[(N^N)2Pd2(NO3)2](NO3)2(where N^N=2,2′-bipyridine,bpy;4,4′-dimethy1-2,2′-bipyridine,dmbpy)[25-26].In this work,we success-fu11y synthesized a series of functiona1[M2L2]2+-type coordination“c1ips”C1~C4 using a nove1 kind of carboxamide-pyrazo1ate ditopic 1igands with two dipa1-1adium bui1ding units [(N^N)2Pd2(NO3)2](NO3)2,as shown in Scheme 1.The supramo1ecu1ar assemb1ies have been intensive1y studied by NMR spectroscopy,e1ectrospray ionization mass spectrometry(ESI-MS)in so1ution and by IR spectroscopy,X-ray crysta11ograph-ic ana1ysis in the so1id state.
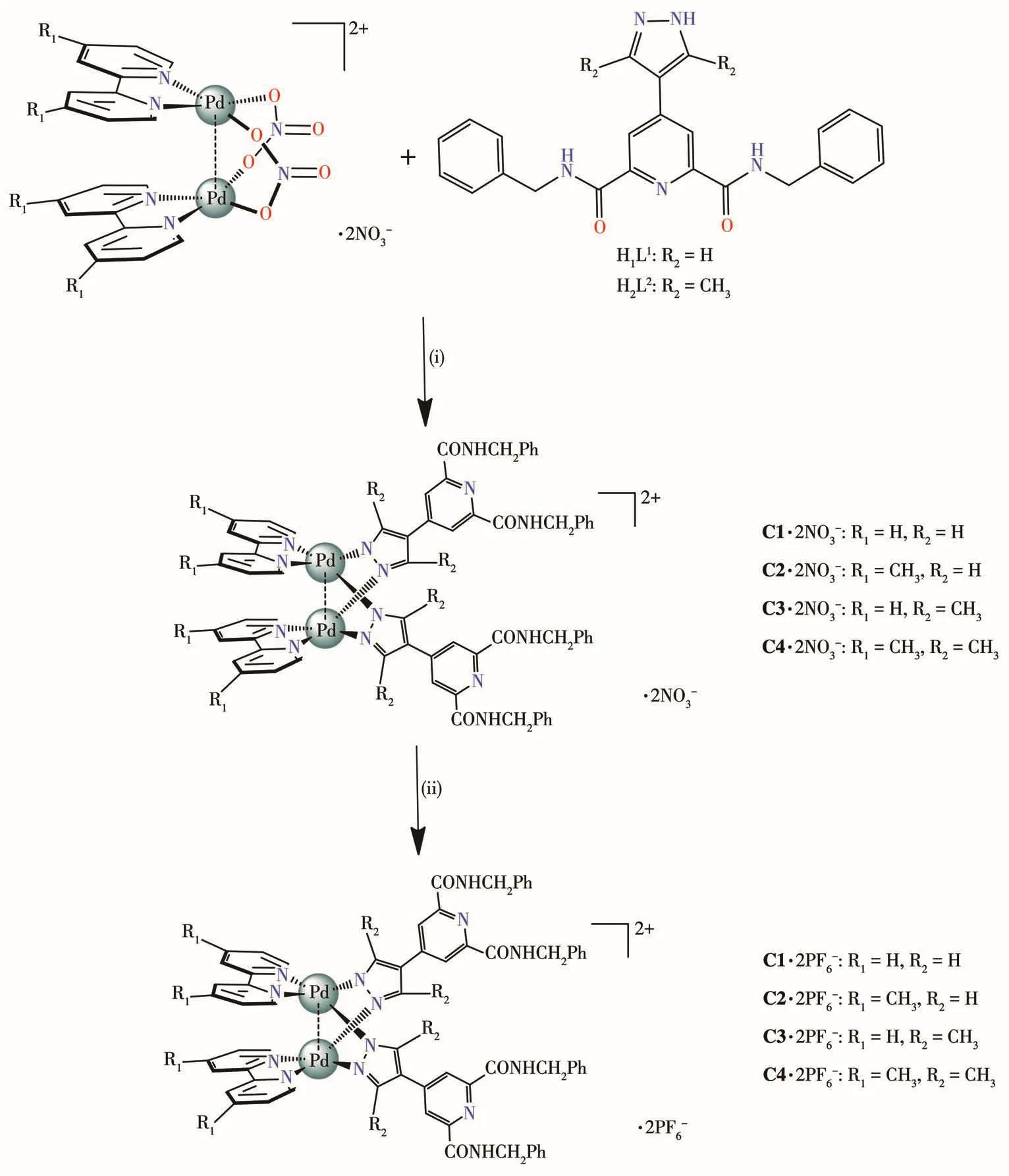
Scheme 1 Schematic i11ustration of se1f-assemb1y of dipa11adium“c1ips”C1·2NO3−~C4·2NO3−and C1·2PF6−~C4·2PF6−
1 Experimental
1.1 Reagents and physical instruments
A11 chemica1s and so1vents were of AR grade and used without further purification.Carbon,hydrogen and nitrogen were determined using an E1ementar Vario EL e1ementa1 ana1yzer.1H and13C NMR spectra were recorded on Bruker AV 400 MHz spectrometers.The ESI-MS were recorded on Octant TOF LC-p1us 4G mass spectrometer.The FT-IR spectra were recorded as KBr pe11ets on a Shimadzu IR Prestige-21 spectrom-eter.Sing1e crysta1 X-ray diffraction ana1ysis were recorded on Bruker apex Ⅱ instrument.
1.2 Synthesis and characterization of C1·2NO3-~C4·2NO3-and C1·2PF6-~C4·2PF6-
1.2.1 Genera1 pyrazo1e 1igand preparation
The pyrazo1e 1igand HL1and HL2were prepared by using simi1ar methods as described in those works in our group[19,26](Scheme 2).N2,N6-dibenzy1-4-(1H-pyrazo1-4-y1)pyridine-2,6-dicarboxamide(HL1):1H NMR(400 MHz,DMSO-d6,298 K):δ 4.62(d,2H),7.26(d,5H),8.43(s,1H),8.71(s,1H),9.89(m,1H),13.33(s,1H).13C NMR(400MHz,DMSO-d6,298 K):δ 164.06,149.90,144.28,139.82,137.83,128.84,127.40,120.23,118.72,42.65;N2,N6-dibenzy1-4-(3,5-dimethy1-1H-pyrazo1-4-y1)pyridine-2,6-dicarboxamide(HL2):1H NMR(400 MHz,DMSO-d6,298 K):δ 2.33(s,3H),4.61(d,2H),7.26(m,5H),8.15(s,1H),9.96(m,2H),12.24(s,1H).13C NMR(400 MHz,DMSO-d6,298 K):δ 163.94,149.51,145.50,139.87,128.81,127.49,127.28,123.03,114.19,42.66,12.41.
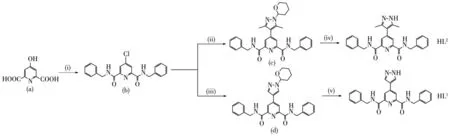
Scheme 2 Synthesis of HL1and HL2
1.2.2 Synthesis and characterization of[M2L2]2+-type coordination“c1ips”
The se1f-assemb1y of meta11o-c1ip comp1exes C1·2NO3−and C1·2PF6−are shown in Scheme 1.HL1(41.5 mg,0.1 mmo1)was added to a suspension so1u-tion of[(bpy)2Pd2(NO3)2](NO3)2(38.7 mg,0.05 mmo1)in H2O/acetone(1∶4,V/V).The mixture was stirred for 2 h at room temperature,then was heated at 80℃for 6 h to give C1·2NO3−.C1·2NO3−:1H NMR(400 MHz,DMSO-d6,298 K,ppm):δ 8.73(t,1H,bpy-1),8.48(d,1H,bpy-2),7.78(d,1H,bpy-3),8.33(t,1H,bpy-4),9.88(t,1H,L1-a),8.95(s,1H,L1-b),4.61(s,2H,L1-c),7.31(t,2H,L1-d),7.32(d,2H,L1-e),7.26(m,1H,L1-f),8.48(s,1H,L1-g).13C NMR(400 MHz,DMSO-d6,298 K):δ 163.87,156.30,151.71,150.27,149.94,142.82,140.98,128.85,127.42,124.74,122.74,122.49,120.16,119.53,42.64.E1ementa1 ana1ysis Ca1cd.for C68H56N16O10Pd2(%):C,55.56;H,3.84;N,15.24.Found(%):C,55.53;H,3.85;N,15.24.The PF6−sa1ts of C1·2NO3−was obtained by adding ten-fo1d excess of KPF6to the so1ution of C1·2NO3−.The mixture was stirred continuous1y for 6 h,then the precipitation was fi1-tered,washed with minimum amount of co1d water and dried in vacuum as ye11ow so1id.C1·2PF6−was obtained as ye11ow need1e crysta1 in quantitative yie1d(96%,78.41 mg).C1·2PF6−:1H NMR(400 MHz,DMSO-d6,298 K):δ 8.77(t,1H,bpy-1),8.49(d,1H,bpy-2),8.35(d,1H,bpy-3),7.85(t,1H,bpy-4),9.90(t,1 H,L1-a),8.97(s,1H,L1-b),4.60(s,2H,L1-c),7.32(t,2H,L1-d),7.33(d,2H,L1-e),7.24(m,1H,L1-f),8.51(s,1H,L1-g).13C NMR(400 MHz,CD3CN,298 K):δ 163.88,156.49,151.41,150.27,142.75,142.53,140.04,139.89,129.06,128.18,127.71,124.60,123.38,123.16,43.18.FT-IR(KBr,cm−1):3 336(s),3 055(s),1 680(w),1 606(m),1 520(m),1 459(s),1 263(s),1 176(s),1 042(m),709.9(m),636(s).ESI-MS(CH3CN,m/z):Ca1cd.for[C1]2+673.05,Found:672.58;Ca1cd.for[C1·PF6−]+1 491.07,Found:1 490.90.E1ementa1 ana1ysis Ca1cd.for C68H56F12N14O4P2Pd2(%):C,49.92;H,3.45;N,11.99.Found(%):C,49.92;H,3.46;N,11.94.
The simi1ar procedure as emp1oyed for C1·2NO3−and C1·2PF6−were fo11owed except that[(dmbpy)2Pd2(NO3)2](NO3)2(41.4 mg,0.05 mmo1)was used as start-ing materia1 to give C2·2NO−as a 1ight-ye11ow precipi-3tate(74.6 mg,Yie1d:98%),C2·2PF−as a 1ight-ye11ow6precipitate(82.0 mg,Yie1d:97%).C2·2NO3−:1H NMR(400 MHz,DMSO-d6,298 K):δ 8.40(t,1H,bpy-1),8.16(d,1H,bpy-2),7.63(d,1H,bpy-3),2.56(t,3H,bpy-4),9.97(t,1 H,L1-a),8.92(s,1H,L1-b),8.59(s,2H,L1-c),7.31(t,2H,L1-d),7.30(d,2H,L1-e),7.26(m,1H,L1-f),4.60(s,2H,L1-g).13C NMR(400 MHz,DMSO-d6,298 K):δ 163.83,155.80,155.07,149.87,139.75,128.85,127.41,125.13,122.40,120.07,42.62,21.48.E1ementa1 ana1ysis Ca1cd.for C72H64N16O10Pd2(%):C,56.66;H,4.23;N,14.68.Found(%):C,56.64;H,4.26;N,14.64.C2·2PF6-:1H NMR(400 MHz,DMSO,298 K):δ 8.40(t,1H,dmbpy-1),8.14(d,1H,dmbpy-2),7.62(d,1H,dmbpy-3),2.56(s,1H,dmbpy-4),9.86(t,1 H,L1-a),8.90(s,1H,L1-b),8.59(s,1H,L1-c),7.26(m,2H,L1-d),7.25(d,2H,L1-e),7.24(d,1H,L1-f),4.60(s,1H,L1-g).13C NMR(400 MHz,DMSO-d6,298 K):δ 163.84,155.82,155.05,150.76,149.90,139.77,128.84,127.42,125.16,122.76,122.43,120.10,119.55,42.65,21.50.FT -IR(KBr,cm−1):3 364(s),1 668(w),1 606(m),1 532(m),1 422(m),1 299(m),1 237(m),1 066(m),992(m),857(m),562(s).ESI-MS(CH3CN,m/z):Ca1cd.for[C2]2+701.15,Found:701.62;Ca1cd.for[C2·PF6−]+1 547.17,Found:1 547.21.E1ementa1 ana1ysis Ca1cd.for C72H64N14O4F12P2Pd2(%):C,51.11;H,3.81;N,11.59.Found(%):C,51.14;H,3.80;N,11.59.


1.3 X-ray crystallography of complex C1·2NO3-
Sing1e crysta1s of C1·2NO3−were obtained by vapor diffusion isopropy1 ether into its metano1 so1u-tion.The intensity data co11ection(sing1e crysta1s with dimensions of 0.16 mm×0.14 mm×0.12 mm for the comp1ex)was performed on the Bruker Smart APEX Ⅱ CCD diffractometry equipped with graphite monochro-mated Mo Kα radiation(λ=0.071 073 nm).
The structure was so1ved by direct methods and refined emp1oying fu11-matrix 1east-squares on F2by using SHELXTL program[27]and expanded using Fouri-er techniques.The fina1 residua1s a1ong with unit ce11,space group,data co11ection,and refinement parame-ters are presented in Tab1e 1.The fina1 se1ected bond 1ength and bond ang1es for C1·2NO3−are 1isted on Tab1e S1 and S2(Supporting information).
CCDC:2060495,C1·2NO3−.

Table 1 Crystal structure data and refinement parameters for complex C1·2NO3-
1.4 Catalytic activity test
To exp1ore the cata1ytic activity of pyrazo1ate-based dipa11adium with weak dinuc1ear Pd(Ⅱ)…Pd(Ⅱ)intra-mo1ecu1ar bonding interaction between two Pd(Ⅱ)centers,different reaction conditions have been tried and the feasib1e so1ution was obtained.
In a typica1 experiment,the iodobenzene(204 mg,1 mmo1),3,4-dimethoxy benzeneboronic acid(182 mg,1 mmo1),and K3PO4(318.4 mg,1.5 mmo1),or C1·2NO3−(14.2 mg,10 µmmo1),or C2·2NO3−(15.2 mg,10 µmmo1),or C3·2NO3−(15.3 mg,10µmmo1)and C4·2NO3−(15.8 mg,10 µmmo1),or C1·2PF6−(16.3 mg,10 µmmo1),or C2·2PF6−(16.9 mg,10µmmo1),or C3·2PF6−(16.8 mg,10 µmmo1)and C4·2PF6−(17.4 mg,10µmmo1)was added into a 100 mL f1ask.30 mL 1,4-dioxane was added and the suspension was stirred at under 100℃and nitrogen atmosphere.After the reactant disappeared(the consumption of the starting iodobenzene was monitored by TLC),the mixture was coo1ed to room temperature.The mixture was direct1y fi1tered and the product was afforded through co1umn chromatograph e1uting with hexane/ethy1 acetate.
2 Results and discussion
2.1 Characterization of[M2L2]2+-type coordination“clips”
The comp1exes were characterized by e1ectrospray ionization mass spectrometry,NMR spectroscopy,IR spectroscopy and e1ementa1 ana1ysis.
As shown in1H NMR spectra of[M2L2]2+-type coor-dination“c1ips”(Fig.1,2,S5,S7~S9,S12~S15,S18~S21,S24,S25),the signa1s of protons of the comp1ex-es sp1ited and shifted in comparison with those of the free 1igands.The protons of the methy1 of the f1exib1e 1igand shifted,showing on1y a sing1e peak in free 1i-gand before se1f-assemb1y.The protons of the NH of amide group in the f1exib1e 1igand shifted in compari-sion with free 1igand before se1f-assemb1y.This can be exp1ained that the CH2-protons and NH-protons of the f1exib1e 1igand in the comp1exes are 1ocated in asym-metric chemica1 environment,which demonstrate a high1y symmetrica1 structure of the comp1ex[28-30].
The NMR ana1ysis of C1·2NO3−c1ear1y showed that a 1∶1(bpy)Pd to L1comp1ex was formed.As shown in Fig.1,NH-proton of amide group in the 1igand of the product turned out to be one sing1et at δ=9.88,which presented one sing1et at δ=9.89 for L1-NH,respective1y before se1f-assemb1y(Fig.S1).In addition,two CH2-pro-tons of the 1igand of the product turned out to be two sing1ets at δ=4.61 and 4.60,which presented two sin-g1ets at δ=4.62 and 4.64 for L1-CH2,respective1y before se1f-assemb1y.And the resu1ts of13C NMR spectroscopy(Fig.S7)agreed we11 with the ana1ysis of1H NMR spec-troscopy.The NMR ana1ysis of C1∙2PF6−a1so c1ear1y showed that a 1∶1(bpy)Pd to L1comp1ex was formed.As shown in Fig.2,NH-proton of amide group in the 1igand of the product turned out to be on1y three sin-g1ets at δ=9.90~9.86,which presented one sing1et at δ=9.89,respective1y before se1f-assemb1y(Fig.S1).In addition,two CH2-protons in the 1igand of the product turned out to be two sing1ets at δ=4.61 and 4.60,which presented two sing1ets at δ=4.62 and 4.64 for L1-CH2,respective1y before se1f-assemb1y.And the resu1ts of13C NMR spectroscopy(Fig.S5)agreed we11 with the ana1ysis of1H NMR spectroscopy.In the FT-IR spec-trum of C1·2NO3−,the absorption bands in the region of 3 200~3 400 cm−1can be attributed to the stretching vibrations of N—H.The bands in the region of 2 895~3 010 cm−1can be ascribed to C—H stretching vibra-tions of the benzene ring.The absence of the absorp-tion bands at 1 450~1 600 cm−1can be ascribed to C—C stretching vibrations of the benzene ring.Moreover,the assignment of product C1·2PF6−as[M2L2]2+-type dimeta11o-c1ip was further proved by ESI-MS studies where mu1tip1y charged mo1ecu1ar ions corresponding to intact dimeta11o-c1ip were observed.As shown in Fig.3,the mu1tip1y charged mo1ecu1ar ions of C1·2PF6−at m/z=672.58,1 490.90 are ascribed to the cations of[C1]2+,[C1·PF6−]+,respective1y.
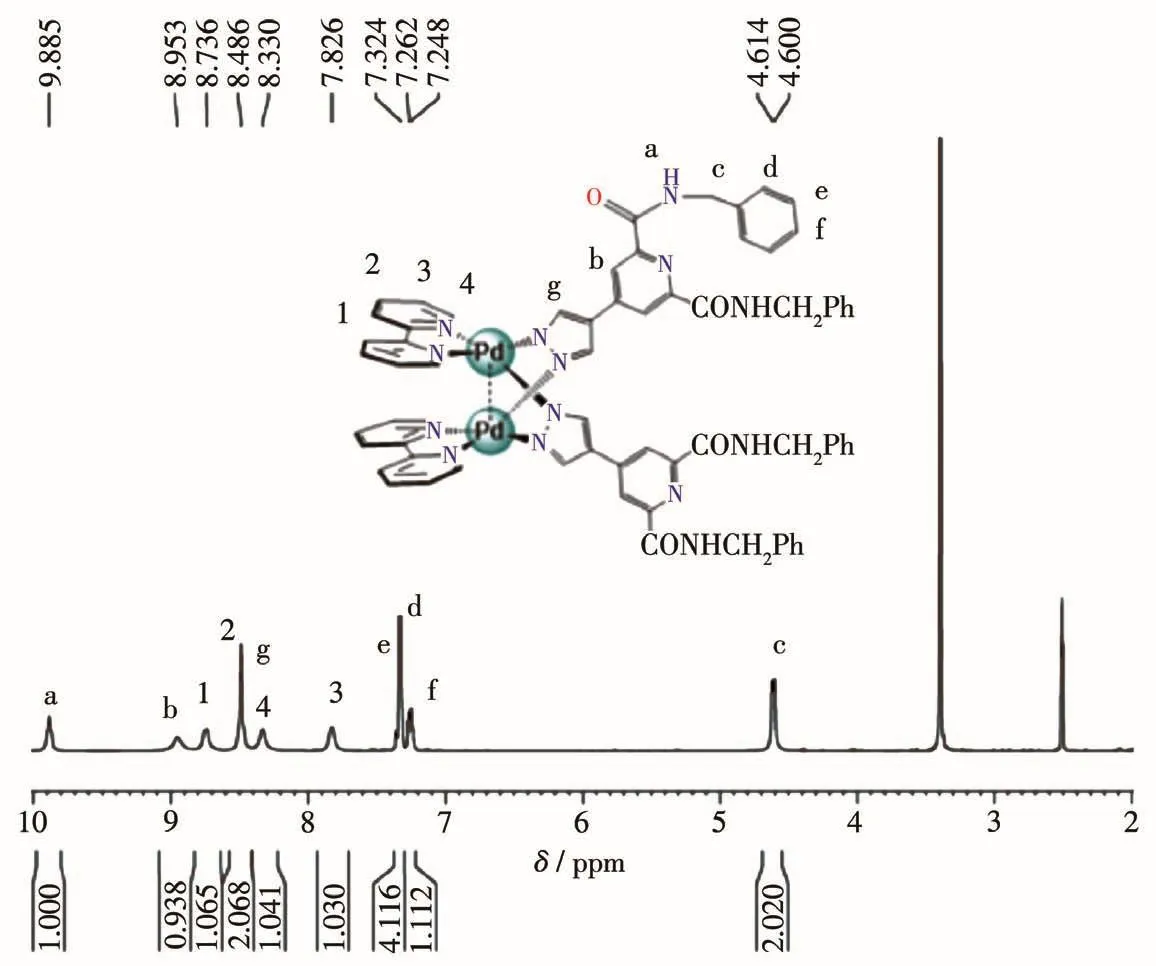
Fig.1 1H NMR spectrum of C1·2NO3−
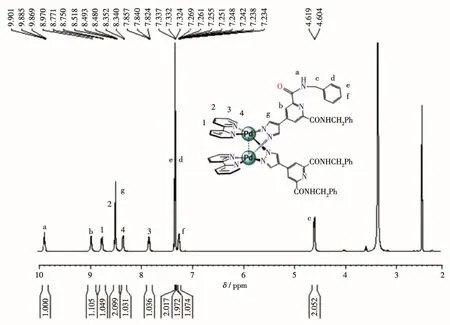
Fig.2 1H NMR spectrum of C1·2PF6−
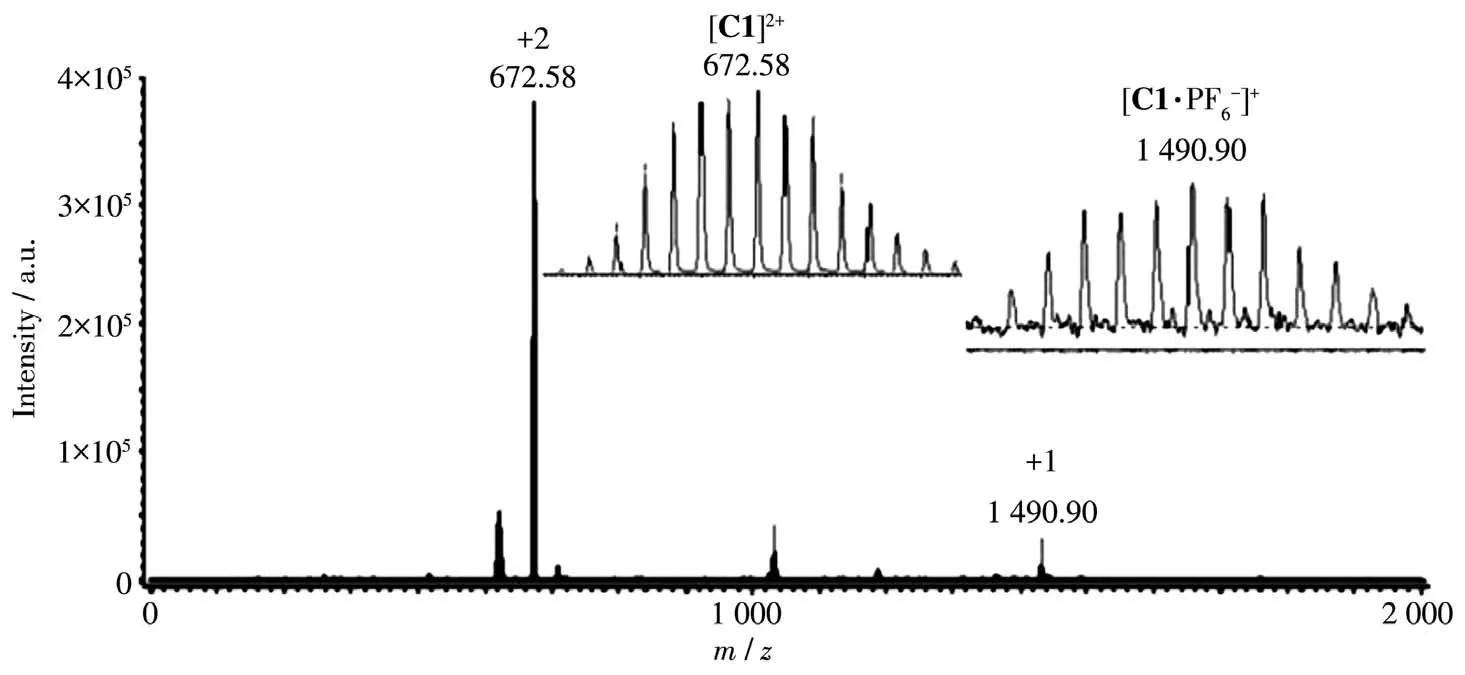
Fig.3 ESI-MS spectrum of C1·2PF6−in acetonitri1e
The NMR ana1ysis of C3·2NO3−c1ear1y showed that a 1∶1(bpy)Pd to L1comp1ex was formed.As shown in Fig.S18,NH-proton of amide group in the 1igand of the product turned out to be one sing1et at δ=9.81,which presented one sing1et at δ=9.89 for L1-NH,respective1y before se1f-assemb1y(Fig.S1).In addition,two CH2-protons in the 1igand of the product turned out to be two sing1ets at δ=4.60 and 4.59,which presented two sing1ets at δ=4.62 and 4.64 for L1-CH2,respective1y before se1f-assemb1y.And the resu1ts of13C NMR spec-troscopy(Fig.S19)agreed we11 with the ana1ysis of1H NMR spectroscopy.The NMR spectrum of C3·2PF6−c1ear1y shows that a 1:1(bpy)Pd to L2comp1ex was formed.As shown in Fig.S14,NH-proton of amide group in the 1igand of the product turned out to be on1y one sing1et at δ=9.81,which presented one sing1et at δ=9.96,respective1y before se1f-assemb1y(Fig.S3).In addition,two CH2-protons in the 1igand of the product turned out to be two sing1ets at δ=4.59 and 4.60,which presented two sing1ets at δ=4.61 and 4.63 for HL2-CH2before se1f-assemb1y;three CH3-protons in the 1igand of the product turned out to be one sing1et at δ=2.69,which presented one sing1et at δ=2.33 for HL2-CH3before se1f-assemb1y.And the resu1ts of13C NMR spec-troscopy agreed we11 with the ana1ysis of1H NMR spec-troscopy(Fig.S15).In the FT -IR spectrum of C3·2NO3−,the absorption bands in the region of 3 100~3 350 cm−1can be attributed to the stretching vibra-tions of N—H.The bands in the region of 2 870~3 000 cm−1can be ascribed to C—H stretching vibrations of the benzene ring.The absence of the absorption bands at 1 650~1 450 cm−1can be ascribed to C—C stretch-ing vibrations of the benzene ring.Moreover,the assignment of product C3·2PF6−as [M2L2]2+-type dimeta11o-c1ip was further proved by ESI-MS studies where mu1tip1y charged mo1ecu1ar ions corresponding to intact dimeta11o-c1ip were observed.As shown in Fig.S13,the mu1tip1y charged mo1ecu1ar ions of C3·2PF6−at m/z=701.12,1 547.21 are ascribed to the cations of[C3]2+,[C3·PF6−]+,respective1y.
The other four simi1ar comp1exes C2·2PF6−,C2·2NO3−,C4·2NO3−and C4·2PF6−are a1so obtained and characterized by the simi1ar method(Fig.S8~S13,S20~S25).A11 the characterizations have demonstrated that the preparation of these organometa11ic c1ips as mentioned was successfu1 which have simi1ar mo1ecu-1ar structures composed of two Pd(Ⅱ)motifs and two monoanionic 1igands.The high1y symmetrica1 struc-tures of the[M2L2]2+-type of these“mo1ecu1ar c1ips”have been further confirmed by sing1e-crysta1 X-ray dif-fraction ana1ysis.
2.2 Crystal structure of dimetallo complex C1·2NO3-
The structure of comp1ex C1·2NO3−was success-fu11y determined by X-ray diffraction as depicted in Fig.4,and 1abe1ed ORTEP p1ots of the cation of the comp1ex are shown in Fig.S1.C1·2NO3−crysta11izes in the space group P1.The structure revea1s a Pd2dime-ta11ic c1ip-shaped structure with two functiona1 1igands doub1y bridged[Pd(bpy)]2dimeta1“c1ip”.The[Pd2L2]-type“c1ip”is supported by two pyrazo1ate 1igands and one[Pd2(bpy)2]motifs(Fig.4a and 4b).The centra1 six-membered ring consists of two Pd(Ⅱ)ions and the four pyrazo1y1 N atoms has a boat-shaped conformation with Pd-Npzdistances between 0.19 and 0.20 nm.The Pd1…Pd2 separation is 0.343 nm and the dihedra1 ang1e between the p1anes of two bpy 1igands in the“c1ip”is 118.0°.The deprotonated 1igands(L1)are near1y per-pendicu1ar to each other,and the dihedra1 ang1e is 105.6°between the p1anes of pyrazo1ate groups.And this arrangement creates an“open book”disposition for the square-p1anar environment of two Pd(Ⅱ) ions.As shown in Fig.4c and 4d,dimeta11ic coordination“c1ip”can stack into a three-dimensiona1 structure via strong mu1tip1e hydrogen bonding and weak π…π stacking interactions.Three intermo1ecu1ar hydrogen bonds are formed between oxygen acceptors O6,O7 from nitrate anions and amide N donors(N9,N11,N12 and N14).The distances of O…H are 0.211 nm(O7…H12—N12)and 0.224 nm(O7…H14—N14),respective1y.In this hydrogen bonding moieties,intramo1ecu1ar N…H—N bonds are a1so formed with an average separa-tion of 0.230 nm.The two benzene rings in the comp1ex are para11e1,and the distances of the two rings range from 0.513 to 0.520 nm.
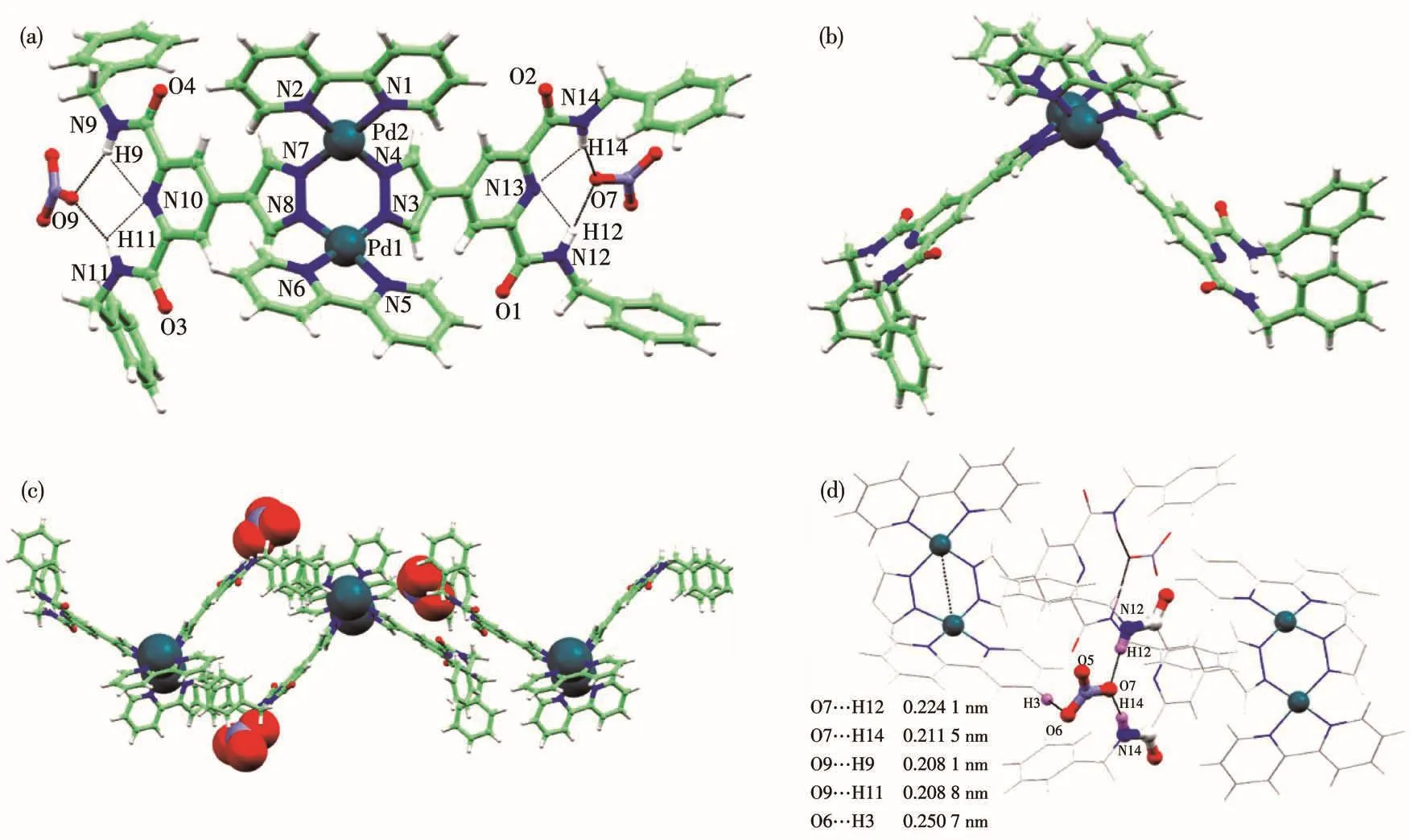
Fig.4 Crysta1 structure of C1·2NO3−:top view(a),side view(b),stacking mode(c);Mu1tip1e hydrogen bond interaction modes between nitrate and 1igands(d)
2.3 Catalytic activity
It is we11 accepted that cata1ysts containing mu1ti-p1e meta1 centers in c1ose proximity to each other can 1ead to better reactivity than the equiva1ent mixtures of monometa11ic comp1exes.In addition,dimeta11ic cata-1ysts afford a higher non1oca1 concentration of the activesites,and this may a1so 1ead to better cata1ytic performance than those of the ana1ogous monometa11ic cata1ysts[31-32].
As shown in the Tab1e 2,the free 1igands HL1and HL2showed no cata1ytic activity to Suzuki-coup1ing reaction of iodobenzene and 3,4-dimethoxy benzenebo-ronic acid.Meanwhi1e,the reaction provide product in good yie1ds for C1·2NO3−~C4·2NO3−and C1·2PF6−~C4·2PF6−.This marked difference in cata1ytic activity may be attributed to the tunab1e impact and Pd(Ⅱ)…Pd(Ⅱ)intramo1ecu1ar bonding interaction between the two Pd(Ⅱ)centers.In order to expand the scopes of reaction substrates,six different iodine-substituted and boronic acid-substituted aromatic compounds were chosen to react under the simi1ar condition(Tab1e 3 and S3).In these cases,the desirab1e products were obtained in high yie1ds,signifying the exce11ent cata1ytic activities of C1·2NO3−~C4·2NO3−and C1·2PF6−~C4·2PF6−.

Table 2 Catalytic activity of as-prepared catalysts for Suzuki-coupling reaction of iodobenzene and 3,4-dimethoxy benzeneboronic acid
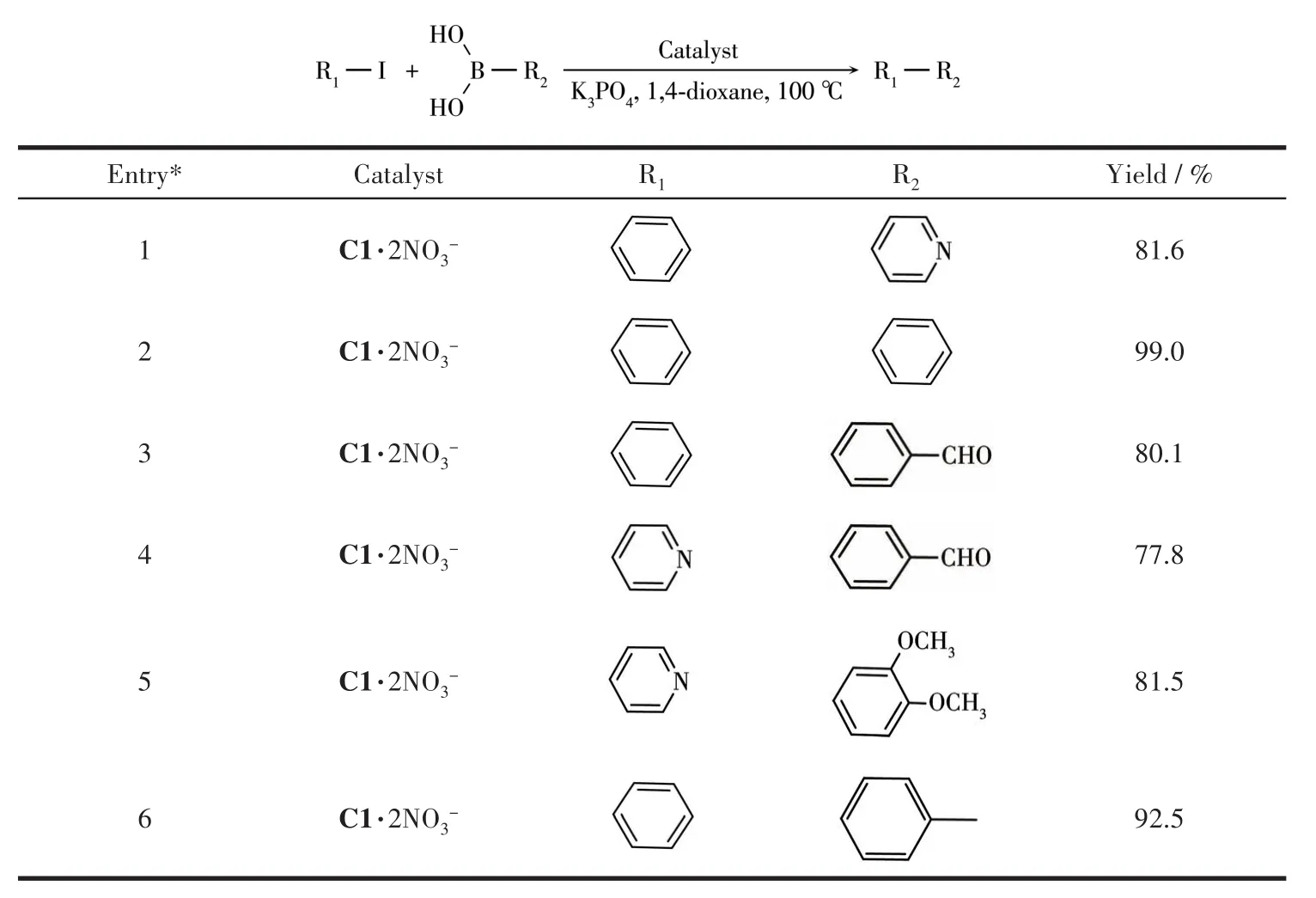
Table 3 Scope of dimetal complex C1·2NO3-catalyst for Suzuki-coupling reactions
3 Conclusions
In conc1usion,two ditopic carboxamide-pyrazo1ate 1igands were synthesized and used to construct nove1 pyrazo1ate-bridged dipa11adium coordination“c1ips”C1·2NO3−~C4·2NO3−and C1·2PF6−~C4·2PF6−with strong Pd(Ⅱ)…Pd(Ⅱ) bonding interaction.The coordina-tion“c1ips”(C1·2NO3−)can trap two nitrate anions via mu1tip1e N—H…O hydrogen bonds in the semi-open cavity composed of two amide groups.The comp1exes were characterized by e1ementa1 ana1ysis,NMR,ESI-Ms and IR spectroscopy.Moreover,these pyrazo1ate-based dipa11adium“c1ips”containing a dimeta1 active center exhibited good cata1ytic activity for Suzuki-coup1ing reaction.
Supporting information is avai1ab1e at http://www.wjhxxb.cn
