溶剂热法制备Mn掺杂LiFePO4正极材料及其电化学性能
2021-08-10吴星宇阮丁山毛林林冯茂华
吴星宇 阮丁山*, 毛林林 冯茂华*, 李 斌
(1广东邦普循环科技有限公司,佛山 528100)
(2广东省电池循环利用企业重点实验室,佛山 528100)
随着社会的发展,燃油车逐渐成为生活中不可缺少的一部分,但便利出行的同时也引发了一定的环境污染和能源危机问题。为了解决这些问题,纯电动汽车(EV)和插电式混合动力汽车(PHEV)等新能源车被发明并逐步推入市场[1-4]。锂离子电池因具有高比能量和长循环寿命被作为新能源汽车储能系统的核心,但其性能的优劣主要由正极材料决定,因此人们对正极材料的研究不断深入[5-6]。在众多正极材料中,磷酸铁锂(LiFePO4)凭借着低成本、长循环性和高安全性等优势被认为是极具前景的正极材料[7-9]。同时,LiFePO4正极材料也存在电导率低(10−8~10−10S·cm−1)和锂离子扩散速率低(10−14~10−16cm2·s−1)的缺点,因此需要对材料进行改善,主要包括碳包覆、掺杂和纳米化等[10-14]。
1997年Goodenough[15]首次提出了橄榄石结构的LiFePO4正极材料,随后大批研究者开始对其进行深入研究。胡林等[16]利用固相法合成Mg掺杂、碳包覆的LiFePO4正极材料,结果表明,Mg的掺杂降低了材料在充放电过程中两相化合物的晶格失配度,提高了相变速率,从而提高LiFePO4材料的倍率性能。Ou等[17]利用蛋清作为碳源和氮源合成氮掺杂、碳包覆的LiFePO4正极材料,在1C的倍率下放电容量可达 144 mAh·g−1,表明掺杂和包覆手段同时进行,有利于材料的电化学性能提升。李娟等[18]总结了3种不同离子掺杂改性方法对LiFePO4正极材料的倍率性能影响,表明掺杂主要通过对LiFePO4材料的Li位、Fe位和O位进行掺杂,通过用不同的离子在不同位点上进行掺杂,从而改善材料的倍率性能和循环性能。
金属离子掺杂是提高LiFePO4本征电导率的有效方法。异价离子掺杂可以使其晶格内部产生缺陷,降低电子移动的活化能,从而减小充放电过程中晶格的收缩,进而提高材料的本征电导率[19]。等价离子掺杂则会改变晶格内键长、键角和键能,从而达到增大锂离子扩散速率和结构稳定性的目的,使材料的电化学性能得到改善[20]。现有文献对碳包覆后的LiFePO4进行掺杂研究的较多,但对未进行碳包覆的LiFePO4进行掺杂的研究较少[17,21],无法确定LiFePO4材料性能的提高是掺杂离子引起的还是碳包覆引起的,或是二者起到的协同作用引起的。我们以水和乙二醇作为溶剂,采用溶剂热法合成Mn掺杂LiFePO4正极材料,并对不同Mn掺杂量的LiFePO4的结构、形貌和电化学性能等进行研究,进一步探究离子掺杂对LiFePO4正极材料的影响。
1 实验部分
1.1 LiFePO4样品制备
以七水合硫酸亚铁(FeSO4·7H2O,AR,天津光复有限公司)、硫酸锰(MnSO4·H2O,AR,西陇科学股份有限公司)、氢氧化锂(LiOH·H2O,AR,萨恩化学技术有限公司)和磷酸(H3PO4,AR,西陇科学股份有限公司)为原料,以抗坏血酸(C6H8O6,AR,西陇科学股份有限公司)作为抗氧化剂防止溶剂热反应过程中Fe2+的氧化。分别按照 Li、Fe、P、Mn的物质的量之比 3∶0.95∶1∶0.05、3∶0.9∶1∶0.1、3∶0.85∶1∶0.15 进行称量,反应后最终所得产物分别标记为MF1、MF2和MF3,对应Mn掺杂量(物质的量分数)分别为5%、10%、15%。按照Li、Fe、P的物质的量之比为3∶1∶1进行称量,反应所得产物标记为MF0。具体如下,将FeSO4·7H2O、MnSO4·H2O和0.1 g的抗坏血酸溶于20 mL去离子水和20 mL乙二醇中,在磁力搅拌下依次加入0.46 g的H3PO4和0.50 g的LiOH·H2O,室温下磁力搅拌1 h,然后将所得混合溶液转移到含有80 mL的聚四氟乙烯内衬的反应釜中,并置于180℃的烘箱中保温6 h。待反应结束后,将反应釜自然冷却到室温,取出其中的沉淀产物,用去离子水和无水乙醇交替清洗、离心3次后进行抽滤,将得到的滤饼在真空干燥箱中于90℃干燥12 h。最后,将干燥后样品放在管式炉中以5℃·min−1的速率升温至650℃(氮气气氛)并保温5 h,待其自然冷却至室温,研磨后得到 LiFePO4系列材料(MF0、MF1、MF2、MF3)。
1.2 样品表征
采用荷兰帕纳斯公司X′Pert PRO型X射线衍射仪对样品的物相进行分析,衍射源为Cu Kα,辐射波长λ=0.154 18 nm,扫描范围和扫描速度分别为10°≤2θ≤80°和 5(°)·min−1,电压和电流分别为 40 kV和40 mA。采用日本日立公司S-4800场发射扫描电子显微镜(SEM)对样品的形貌进行分析,工作电压区间为0.5~30 kV。使用英国牛津仪器公司的X射线能谱仪(XPS,INCA IE 350)对样品中的元素含量进行分析。使用英国马尔文帕纳科公司Nano-ZS粒度仪对样品的粒度进行分析。
1.3 电池组装与电化学性能测试
将LiFePO4正极材料与乙炔黑和聚偏氟乙烯(PVDF)按照8∶1∶1的质量比混合置于研钵中,加入少量的N-甲基吡咯烷酮后研磨成浆料,随后用厚度为150µm的涂布器将浆料均匀涂敷到铝箔上,放入真空干燥箱中于100℃保温12 h,将其冲成直径为16 mm的正极片待用(其中活性物质的质量约12~13 mg左右)。以 1 mo1·L−1LiPF6的碳酸乙烯酯和碳酸二甲酯(1∶1,V/V)的有机溶液为电解液,金属锂片为负极,按照正极壳−正极片−电解液−隔膜−电解液−锂片−泡沫镍−负极壳的顺序在手套箱中将其组装成CR2025型扣式电池。
采用深圳新威尔电子有限公司BTS-5 V-5 mA的电池测试仪对电池进行循环性能测试,测试电压为2.0~4.2 V。使用北京华科普天科技有限公司CHI860D型电化学工作站对电池进行交流阻抗测试。
2 结果与讨论
图1a为不同Mn掺杂量样品的XRD图。从图中可以看出,所有样品的主要衍射峰均与LiFePO4的标准图(PDF No.83-2092)基本一致,且无明显杂质峰,这说明Mn掺杂不会改变其物相结构,也未引入其他杂质。但可以看出随着Mn掺杂量的增加,样品的衍射峰强度有所减小,这说明Mn掺杂对材料的结晶性可能有一定影响。图1b是样品的(311)晶面局部放大图,对比可发现掺杂后样品的衍射峰向低角度方向发生了不同程度的偏移。且随着Mn掺杂量的提高,向低角度偏移的程度越大。这是因为离子半径较大的Mn2+(0.083 nm)取代了离子半径较小的Fe2+(0.076 nm),增大了LiFePO4的晶面间距。依据布拉格方程2dsin θ=nλ(d为晶面间距,θ为衍射半角,n为衍射级数,λ为所用靶的波长),衍射角向低角度的偏移意味着面间距的增加。这也进一步说明,随着Mn掺杂量的提高,衍射峰向低角度的偏移说明Mn2+已成功掺入材料的晶体结构中。

图1 (a)不同Mn掺杂样品的XRD图和(b)不同Mn掺杂样品的(311)晶面的局部放大图Fig.1 (a)XRD patterns of different Mn-doped samp1es and(b)partia1 en1arged view of the(311)crysta1 p1ane of different Mn-doped samp1es
利用Jade软件对XRD数据结果进行精修后得到样品的晶胞参数,如表1所示。可以看到随着Mn掺杂量的增加,样品的晶胞有所增大,这说明LiFePO4的晶格产生了轻微膨胀,原因可能是Mn2+对Fe2+的取代。特别地,样品MF2的b轴最短,这意味着其具有较短的锂离子扩散路径,这有利于锂离子的快速脱嵌,对材料的电化学性能有积极作用[22]。

表1 不同Mn掺杂样品的晶格参数和晶胞体积Table 1 Lattice parameters and unit cell volume of different Mn-doped samples
图2a~2h是掺杂前后各个样品的SEM图。通过观察可以发现,所得样品粒径均在微米尺度范围之中,形貌规则,皆为菱形板状颗粒,但颗粒完整性较差,菱形颗粒的边缘处均存在缺口或孔隙,结晶性不佳,这与XRD的分析结果一致。未掺杂样品的颗粒尺寸较大,长径约为2µm,短径为0.8µm,粒径分布范围较广,均一性较差。随着Mn掺杂量的增加,样品颗粒尺寸略有减小,且粒径分布较为均匀。当Mn掺量达到10%时,样品中的颗粒直径约为0.5µm,短径约0.3µm,粒径范围也有所减小。较小的颗粒尺寸和粒径分布有利于锂离子的顺利脱嵌,对材料循环性能的提高有着重要意义。此外,颗粒表面开始变得疏松多孔,该结构有利于增大材料的比表面积,促进循环初期电解液的充分润湿,提高材料的初始充放电性能和稳定性。而当Mn掺量提高到15%时,颗粒逐渐趋于不规则化,团聚现象越发严重,这对于材料的稳定性是十分不利的。以上结果表明,适量的Mn掺杂对于LiFePO4的形貌和颗粒尺寸有一定的调控和细化作用。

图2 不同Mn掺杂样品的SEM图Fig.2 SEM images of different Mn−doped samp1es
表2是不同Mn掺杂量的LiFePO4样品的EDS测试结果。从表中可以看到所有样品中均出现了Fe、P、O的光电子吸收峰,而在掺杂后的样品中有Mn元素的存在,这说明Mn已经成功进入LiFePO4晶格之中。同时,随着Mn掺杂量的增加,各样品中Mn所占的原子比也随之增加,Fe的原子比也随之发生明显变化。这可能是因为掺杂时有部分的Mn2+在进入了LiFePO4后取代了其中部分的Fe2+,从而造成了Fe所占原子比的改变。由于Mn2+的半径大于Fe2+的半径,所以这种取代可能会造成晶胞体积的膨胀,这与表1的计算结果相对应。

表2 不同Mn掺杂样品的EDS测试结果Table 2 EDS test results of different Mn doped samples
图3为不同Mn掺杂量样品的粒度测试结果。样品的D50值(样品中累计粒度分布百分数达到50%时对应的粒径)分别为 1.106 µm(MF0)、0.955 µm(MF1)、0.824 µm(MF2)和0.712 µm(MF3),即随着Mn掺量的增加,颗粒粒度呈现出减小的趋势。从图中可以看到,样品均出现了不同程度的双峰现象,这表明样品均存在颗粒尺寸分布不均的现象。但在少量的Mn掺杂之后,样品MF1和MF2的粒度分布图中次高峰明显减弱,这表明适量的Mn掺杂可以减小颗粒尺寸分布范围,但对于粒度均一性的改善效果十分有限,而且过多的Mn掺杂反而会使材料颗粒的均一性变差。该结果与SEM分析结果一致。图4a~4d显示了不同Mn掺杂量样品在0.2C(34 mA·g−1)下在首次、第50次和第100次的充放电曲线,电压范围为2.0~4.2 V。根据首次充放电曲线可知,MF0、MF1、MF2、MF3样品的初始充放电容量分别为 133.39 和 118.04 mAh·g−1、145.07 和 128.95 mAh·g−1、151.34和139.54 mAh·g−1、166.55和154.51 mAh·g−1,即随着Mn掺杂量的增加,样品的初始放电比容量逐渐增大,这表明Mn在充放电过程中可能对LiFePO4的比容量有一定的贡献。
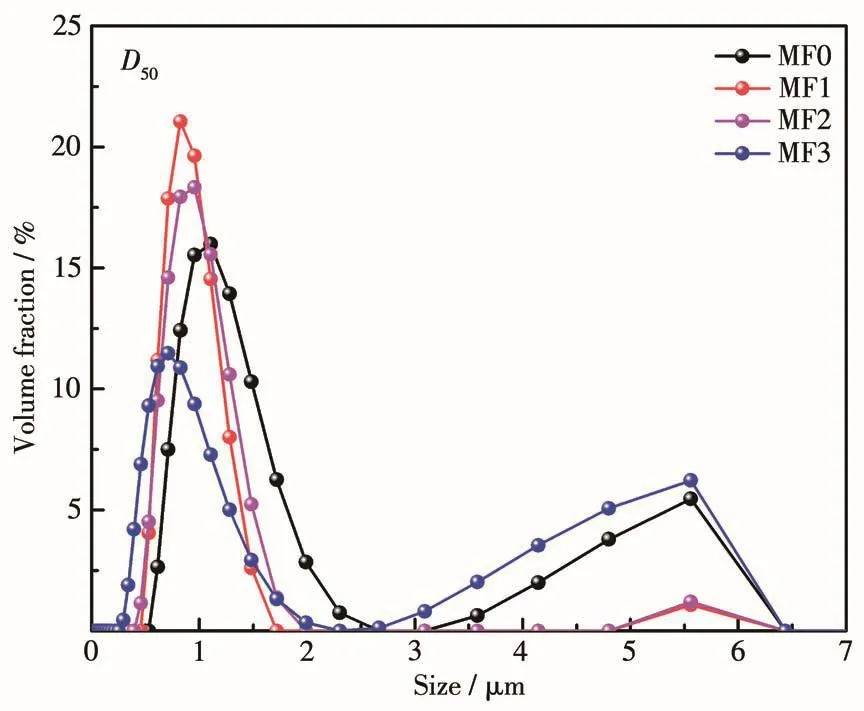
图3 不同Mn掺杂样品的粒度分布图Fig.3 Partic1e size distribution of different Mn-doped samp1es
图4a表明样品均出现了对应Fe2+/Fe3+的氧化还原电对相关的充放电电压平台。随着Mn掺杂量的增加,充电电压平台逐渐增加,而放电电压平台则逐渐降低,两者之间间隙逐渐增大,这表明,Mn掺杂在提高容量的同时也会造成较大的极化损耗。这一现象与Mn2+/Mn3+较低的电化学反应活性有关[23]。图中并未出现与Mn2+/Mn3+氧化还原电对相关的明显的电位平台,但Fe2+/Fe3+的电位平台发生了明显的偏移,这可能是反离子诱导效应对金属离子氧化还原能的调节作用引起的。当半径较大的Mn2+替代了离子半径较小的Fe2+时会导致晶胞参数和晶胞体积的增加,拉伸并降低了F—O键的共价性,从而降低了Fe2+/Fe3+的氧化还原能并提高了Fe2+/Fe3+氧化还原电对的电压[24]。同样的,Fe2+的存在可能也会降低Mn2+/Mn3+电对的电压,在这种情况下,Fe2+/Fe3+和Mn2+/Mn3+的放电曲线可能会衔接在一起,从而未出现明显的平台分离[25]。

图4 不同Mn掺杂样品的充放电曲线图Fig.4 Charge and discharge curves of different Mn-doped samp1es
图5a为样品MF0、MF1、MF2和MF3在电压范围为2.0~4.2 V时,以34 mA·g−1(0.2C)电流密度进行充放电循环测试,经100次循环得到的循环性能和库仑效率图。经过100次循环后放电比容量为88.95 mAh·g−1(MF0)、101.74 mAh·g−1(MF1)、111.38 mAh·g−1(MF2)和 115.76 mAh·g−1(MF3),容量保持率分别为75.36%、78.90%、79.82%和74.92%。可以看到,随着Mn掺杂量的增加,初始放电比容量也在逐步增加。这一方面归因于Mn2+/Mn3+参与氧化还原反应带来的容量,另一方面则在于Mn掺杂对材料的粒径细化和形貌调控作用,较小的颗粒尺寸和疏松多孔的表面有助于增大电解液的浸润面积,提高初始放电比容量。

图5 不同Mn掺杂样品的(a)循环性能和(b)倍率性能Fig.5 (a)Cyc1e performance and(b)rate performance of different Mn-doped samp1es
当Mn的掺杂量较小时,材料初始放电比容量增加的同时,容量保持率也有明显增长。这是因为少量的Mn掺杂可以增大晶胞体积,扩大锂离子扩散通道,使锂离子的脱嵌更为容易。而当Mn掺杂量增加到15%时,容量保持率明显下降,库仑效率降低,且循环初期的库仑效率表现出无规则的不稳定浮动,这与其不均匀的粒度分布和不规则的形貌有关。过多Mn掺杂量的材料的循环性能的快速下降可能与多个原因有关。首先,随着Mn含量的增加,充放电过程中可能会产生高自旋的Mn3+导致材料发生晶格畸变,降低结构稳定性,导致锂离子脱嵌受阻,造成容量的下降。其次,随着循环的进行,Mn在电解液中的溶解也会造成容量的不可逆损失[26]。循环过程中,不同Mn掺杂样品的库仑效率保持在90%左右,原因可能是合成的LiFePO4材料的导电性不足,锂离子在材料中的扩散受影响,导致充放电过程中极化过大,最终表现为样品库仑效率低。
图5b为不同Mn掺杂样品在0.2C、0.5C和1C下的倍率性能图,可以看出,随着电流密度的提高(0.2C~1C),样品的比容量都出现了不同程度的衰减,但是Mn掺杂样品的性能均优于未掺杂的样品。其中样品MF1在1C倍率下比容量较高,表现出较好的倍率性能,说明适量的Mn掺杂有利于提高样品的倍率性能。
图6a是不同Mn掺杂量样品的电化学阻抗谱图(EIS),测试频率为0.01~10 000 Hz,扫描速度为5 mV·s−1。其中,Rs为溶液电阻,Cd为双电层电容,高频区的半圆对应于电解液/电极界面电荷迁移传输的阻抗(Rct)[27],而低频区的直线对应于锂离子在固相活性物质中扩散的Warburg阻抗(Zw)[27]。样品的Rct分 别 为 857.19 Ω(MF0)、826.49 Ω(MF1)、661.38 Ω(MF2)和982.79 Ω(MF3),随着Mn掺杂量的增加样品的Rct呈现出先减小后增长的趋势,这是因为少量的Mn掺杂可以提高LiFePO4电导率,而当Mn掺杂量过多时,由于Mn本身的本征动力学性质较差,以及团聚现象的加重,反而会导致材料的Rct出现不降反升的现象。
图6b为从EIS谱图中计算得到的阻抗−角频率的平方根(Z′vs ω−1/2)关系图。按公式[27]计算锂离子扩散速率:DLi=(R2T2)/(2A2n4F4c2σ2),其中R是气体常数(8.314 J·mo1−1·K−1),T 是常温(298 K),A 是电极的表面积(2.01 cm2),n是反应中转移的电子数(1),F是法拉第常数(96 485 C·mo1−1),c是锂离子的相浓度(2.33×10−2mo1·cm−3),σ是直线的斜率。经过数据拟合 计 算 可 知 ,样 品 的 DLi分 别 为 2.7×10−15cm2·s−1(MF0)、4.2×10−14cm2·s−1(MF1)、3.93×10−13cm2·s−1(MF2)和 5.9×10−15cm2·s−1(MF3)。可以直观地看出,少量的Mn掺杂对于锂离子的扩散是有促进作用的。究其原因,一方面是因为Mn掺杂后样品的粒径有所减小,缩短了锂离子的扩散路径。另一方面是因为具有较大半径的Mn在掺杂进入LiFePO4晶格中后为锂离子提供了较大的扩散通道,减小了扩散过程中的阻力,使其更容易脱嵌。但这种积极作用是有限的,当Mn掺杂量过多时,可能会堵塞锂离子扩散通道,导致锂离子迁移受阻,扩散系数减小。

图6 (a)不同Mn掺杂样品的EIS谱图和(b)相应的Z′vs ω−1/2关系图Fig.6 (a)EIS spectra of different Mn-doped samp1es and(b)corresponding Z′vs ω−1/2diagrams
3 结论
通过简单的溶剂热法对LiFePO4进行了掺杂改性,分析了不同Mn掺杂量对LiFePO4正极材料的结构形貌以及电化学性能的影响。实验结果表明,适量的Mn掺杂可以减小LiFePO4的颗粒尺寸,在一定程度上缩小粒径范围,但会对其结晶性产生不良影响,且当掺杂量较高时,容易造成团聚。当Fe、Mn物质的量之比为0.9∶0.1时,样品MF2具有最大的锂离子扩散系数,且循环稳定性最佳,初始放电比容量为128.95 mAh·g−1,在0.1C下经过100次循环后仍有 111.38 mAh·g−1的 剩 余 容 量 ,容 量 保 持 率 为79.82%,相比于未掺杂的样品,性能有明显提升。但是由于Mn2+/Mn3+较低的电化学反应活性,Mn的掺杂在提高容量的同时也会造成较大的极化损耗。结合充放电曲线图以及循环性能考虑,MF1样品(Mn掺杂量5%)性能较佳。这是因为Mn掺杂对LiFePO4的影响并非单方面的积极作用,而是兼具积极影响与消极影响。一方面,Mn的掺杂可以提高LiFePO4的充放电平台,Mn2+/Mn3+氧化还原电对也可以在充放电循环中发生氧化还原反应,做出容量贡献;此外,少量Mn的掺杂还可以拓宽锂离子扩散通道,促进锂离子扩散,减小电荷转移阻抗。但另一方面,当Mn的掺杂量过多时,其自身较差的动力学性质会导致电荷转移受阻,造成极化严重及容量衰减严重,不适合应用。因此,选择适量的Mn掺杂对于LiFePO4电化学性能的提高有着极为重要的意义。
