半夹芯16电子碳硼烷化合物Cp*CoS2C2B10H10与含磷化合物的反应性
2021-08-10叶红德张昕瑜朱清嵘何橹灵彭化南刘紫薇
叶红德 张昕瑜 肖 欣 朱清嵘 何橹灵 彭化南*, 刘紫薇
(1上饶师范学院化学与环境科学学院,江西省普通高校应用有机化学重点实验室,上饶 334001)
(2海南师范大学,海南省热带药用植物化学重点实验室,海口 571158)
二十面体闭式−碳硼烷(C2B10H12)根据分子中2个碳原子的相对位置可以分为3种异构体:1,2-C2B10H12、1,7-C2B10H12和 1,12-C2B10H12(即 o-碳硼烷、m-碳硼烷和p-碳硼烷)。近年来,这些二十面体闭式−碳硼烷由于它们独特的3D拟芳香性几何结构和推−拉电子特性而得到了广泛的研究[1],含碳硼烷结构单元材料的光物理性质吸引了越来越多的研究兴趣[2]。人们通过把碳硼烷结构单元引入到发光材料中以便提高和改善其发光性能[3],如把碳硼烷作为有效的构造材料用在有机和聚合物领域以便获得优异的发光和电子材料[4],或把碳硼烷结构单元引入荧光有机体系[5]、聚合物[6]和金属配合物[7]。实验结果显示,碳硼烷在协调光物理性质方面起着独特的作用。不过,就含碳硼烷结构单元材料的光物理性质而言,碳硼烷笼体本身的作用仍然需要进一步进行研究,对于o-碳硼烷衍生物更应如此,因为o-碳硼烷衍生物拥有一个独特的C—C键(键距范围在0.162~0.215 nm)[8],并且通常能使荧光猝灭[9]。
近年来,人们对含钴金属离子配合物的紫外和荧光性能[10-14]进行了广泛研究。但据我们所知,目前还没有对含有钴离子的碳硼烷衍生物的荧光和紫外性能进行测试。我们首先合成含有钴离子的半夹芯式16电子碳硼烷化合物Cp*CoS2C2B10H10,再用合成的Cp*CoS2C2B10H10分别与二苯基甲基膦、苯基二甲基膦和三甲基膦反应生成含有碳硼烷笼体和钴离子的加成物,最后对这些加成物进行紫外和荧光性能测试。
1 实验部分
1.1 试剂与仪器
所有化学试剂均为分析纯,未做进一步纯化处理直接使用。溶剂在氮气气氛下用金属钠(石油醚、乙醚和四氢呋喃)或者氢化钙(二氯甲烷)干燥,使用前进行重蒸。正丁基锂(2.0 mo1·L−1in cyc1ohexane,A1drich)直接使用。Cp*CoS2C2B10H10参照相关文献[15-17]合成。合成过程采用标准的Sch1enk技术。使用Bruker SMART Apex Ⅱ型X射线单晶衍射仪收集单晶数据。采用Thermo Fisher Scientific质谱仪进行质谱测试,Nico1et 6700型FTIR红外光谱仪测定红外光谱,Purkinje TU-1901型紫外分光光度计测定紫外可见(UV-Vis)光谱,F-7000型荧光光谱仪测定荧光光谱。
1.2 化合物1~3的合成
在氩气保护下把Cp*CoS2C2B10H10(80.1 mg,0.2 mmo1)和二苯基甲基膦(100.1 mg,0.5 mmo1)加入到20 mL二氯甲烷中,室温反应10 h。之后减压抽去溶剂,粗产物经200~300目硅胶柱层析分离,洗脱剂为石油醚/CH2C12(2∶1,V/V),得到产物1。产物2、3用类似方法合成得到(Scheme 1)。
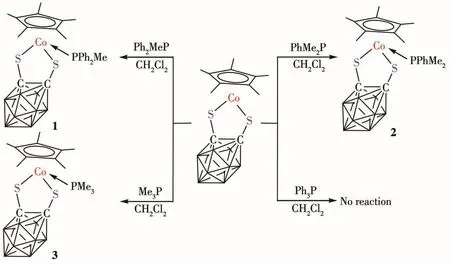
Scheme 1 Synthesis of 1~3
1:产率:95%(114.2 mg)。熔点:298 ℃(分解)。1H NMR(CDC13):δ 7.54~7.36(m,10H,Ph-H),2.05(s,3HCH3),1.60(s,15H,Cp*)。13C NMR(CDC13):δ 133.27(Ph-CH),130.50(Ph-C),128.22(Ph-CH),128.13(Ph-CH),94.51(Cp*-Cring),92.09(carborane-C),77.20(carborane-C),10.42(Cp* -CH3),9.95(CH3)。11B NMR(CDC13):δ−1.64(4B),−4.52(3B),−7.58(3B)。ESI-MS(positive mode,CH2C12/CH3OH,5∶1,V/V):m/z=639.18[C25H38B10CoS2PK]+。IR(KBr,cm−1):2 572(B—H)。元素分析按C25H38B10CoPS2计算值(%):C,49.98;H,6.38。实测值(%):C,50.02;H,6.35。
2:产率:96%(103.4 mg)。熔点:285 ℃(分解)。1H NMR(CDC13):δ 7.54~7.39(m,5H,Ph-H),1.71(s,3H,CH3),1.68(s,3H,CH3),1.60(s,15H,Cp*)。13C NMR(CDC13):δ 134.64(Ph-CH),130.58(Ph-C),130.45(Ph-CH),128.52(Ph-CH),96.97(Cp*-Cring),95.04(carborane-C),77.20(carborane-C),16.49(CH3),16.13(CH3),9.65(Cp*-CH3)。11B NMR(CDC13):δ−1.78(3B),−5.36(3B),−7.20(4B)。ESI-MS(positive mode,CH2C12/CH3OH,5∶1,V/V):m/z=577.19[C20H36B10CoS2PK]+。IR(KBr,cm−1):2 579(B—H)。元素分析按C20H36B10CoS2P计算值(%):C,44.60;H,6.74。实测值(%):C,44.55;H,6.78。
3:产率:97%(94.5 mg)。熔点:270 ℃(分解)。1H NMR(CDC13):δ 1.60(s,15H,Cp*),1.51(s,3H,CH3),1.50(s,3H,CH3),1.49(s,3H,CH3)。13C NMR(CDC13):δ 97.04(Cp*-Cring),95.08(carborane-C),92.05(carborane-C),16.65(CH3),10.01(Cp*-CH3)。11B NMR(CDC13):δ−1.69(2B),−5.56(4B),−9.02(4B)。 ESI-MS(negative mode,CH3OH):m/z=585.06[C15H34B10CoS2P+DMSO+CH3O]−。IR(KBr,cm−1):2 573(B—H)。元素分析按C15H34B10CoS2P计算值(%):C,37.80;H,7.19。实测值(%):C,37.75;H,7.16。
1.3 晶体结构测定
分别选取大小尺寸为0.20 mm×0.15 mm×0.12 mm(1)、0.20 mm×0.15 mm×0.12 mm(2)、0.20 mm×0.18 mm×0.12 mm(3)的单晶,置于 Bruker SMART Apex Ⅱ型X射线单晶衍射仪上进行衍射实验,分别在222(2)、296(2)和296(2)K下,用石墨单色化的Mo Kα(λ=0.071 073 nm)射线,采用ω-2θ扫描方式收集衍射数据。衍射数据用SAINT程序进行还原处理,用SADABS程序进行吸收校正。全部数据经Lp校正和吸收校正,用SHELXS-97[18]进行晶体结构解析,用SHELXL-97[19]进行结构精修。化合物1、2和3的有关晶体学数据详见表1,部分键长和键角数据列于表2。
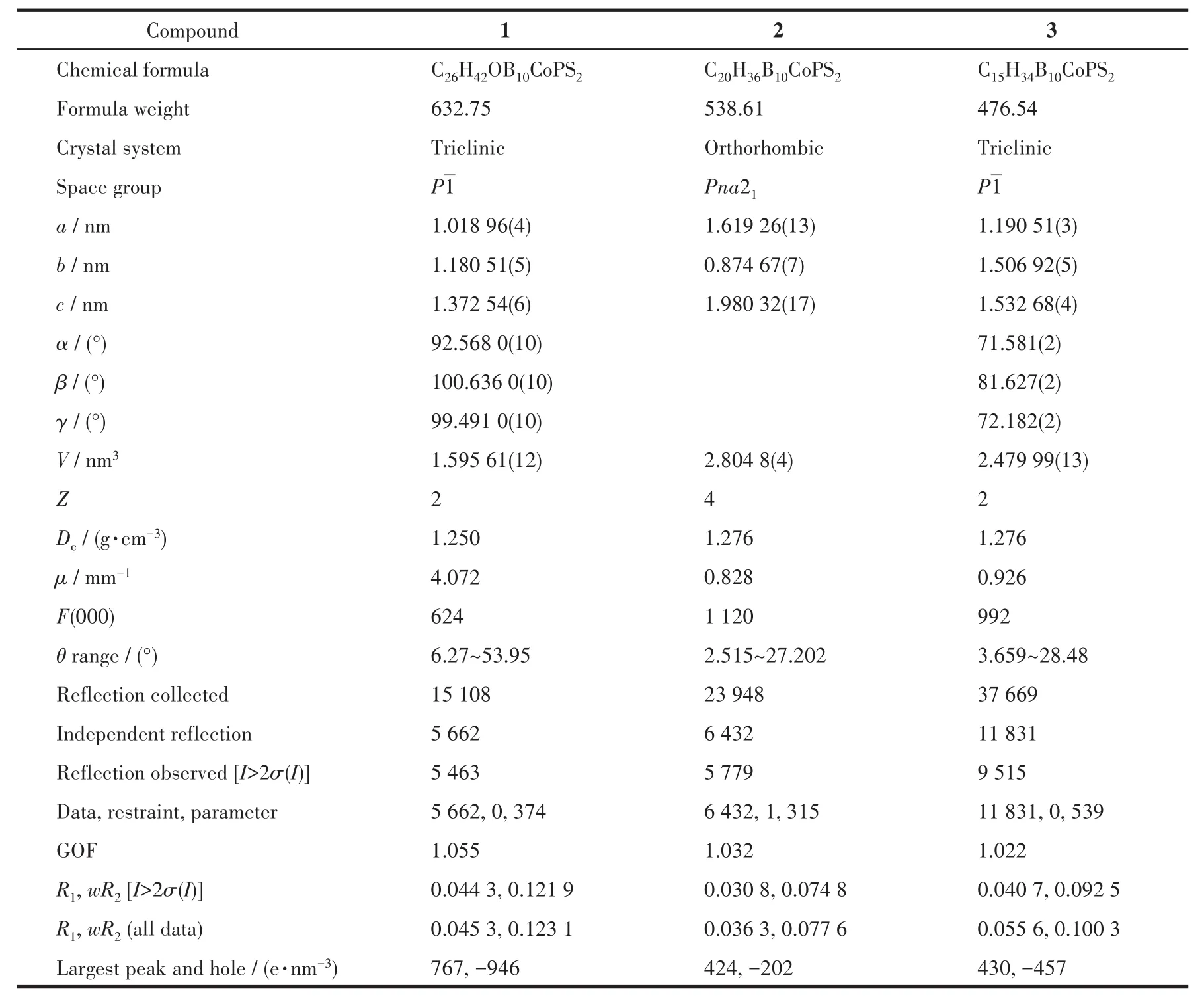
表1 化合物1~3的晶体和结构精修数据Table 1 Crystal and structure refinement data for compounds 1~3
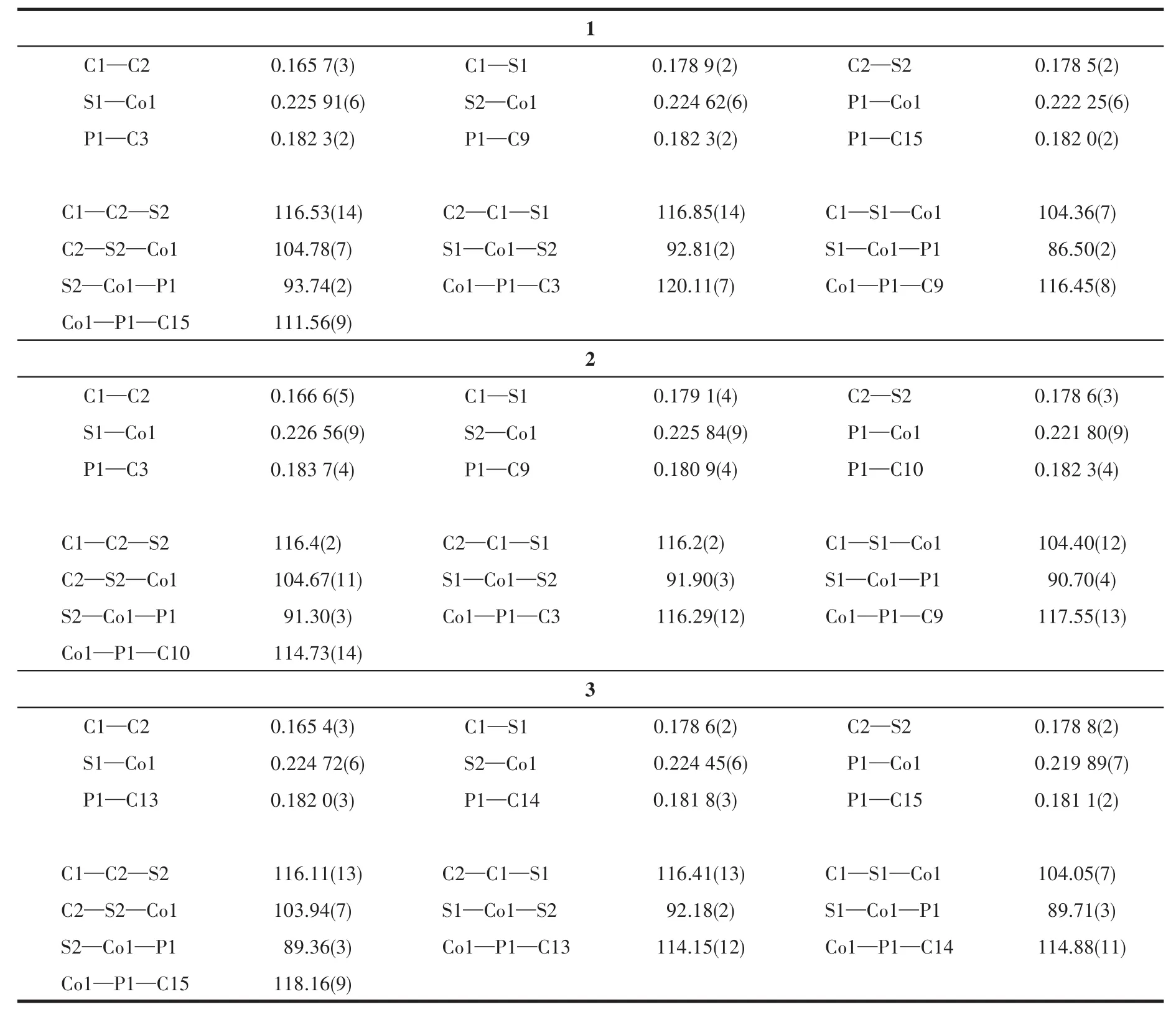
表2 化合物1~3的部分键长(nm)和键角(°)Table 2 Selected bond lengths(nm)and bond angles(°)for compounds 1~3
CCDC:2056803,1;2056804,2;2056805,3。
2 结果与讨论
2.1 化合物的结构
化合物1~3是半夹芯16电子碳硼烷化合物Cp*CoS2C2B10H10分别和二苯基甲基膦、苯基二甲基膦和三甲基膦发生加成反应的产物。从化合物1的分子结构图(图1)可以看出,与金属中心钴离子配位的辅助配体是Cp*基团。C1、C2、S1、S2和Co1五个原子组成了一个五元环。由于二苯基甲基膦分子中的磷原子与原料Cp*CoS2C2B10H10分子中的钴离子配位,导致该五元环不再是一个平面,以S1…S2为矢量的二面角是159.5°。由于磷原子提供一对电子与钴离子配位,钴离子核外最外层电子数达到18个电子的稳定结构。P—Co键长为0.222 25(6)nm,属于正常的P—Co键距离范围[20]。1的光谱和分析测试数据和它的固体结构一致。例如,核磁共振氢谱1H NMR数据显示,化学位移为7.54~7.36的多重峰是2个苯环上10个氢原子的吸收峰,化学位移为2.05的单峰是与磷原子相连的甲基吸收峰,化学位移为1.60的单峰是Cp*环上的5个甲基吸收峰。另外,1的碳谱、硼谱、质谱、元素分析和红外光谱等表征数据与1的结构都相符合。
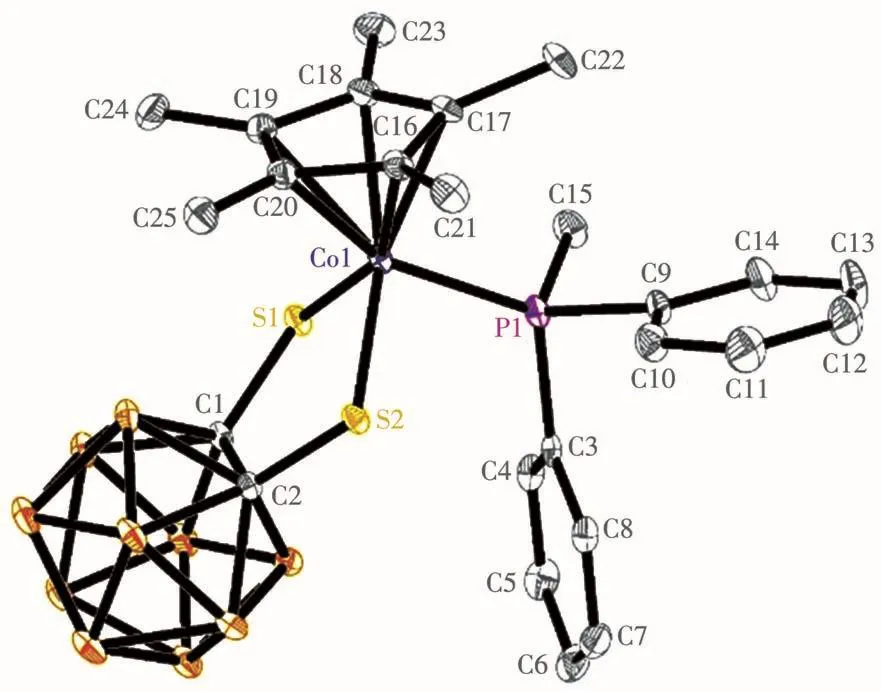
图1 化合物1的椭球几率为30%的分子结构图Fig.1 Mo1ecu1ar structure of compound 1 with therma1 e11ipsoids at 30% probabi1ity 1eve1
半夹芯16电子碳硼烷化合物Cp*CoS2C2B10H10和苯基二甲基膦发生加成反应得到产物2。2的分子结构与1的分子结构类似(图2),同样由于苯基二甲基膦分子中的磷原子与钴离子配位,使分子中C1、C2、S1、S2和Co1五个原子组成的五元环不是一个平面,以S1…S2为矢量的二面角是156.0°。2的光谱和分析测试数据和它的固体结构一致。例如,核磁共振氢谱1H NMR数据显示,化学位移为1.71和1.68的单峰是与磷原子相连的2个甲基吸收峰,化学位移为1.60的单峰是Cp*环上的5个甲基吸收峰。同样,2的碳谱、硼谱、质谱、元素分析和红外光谱等表征数据与2的结构也都相符合。
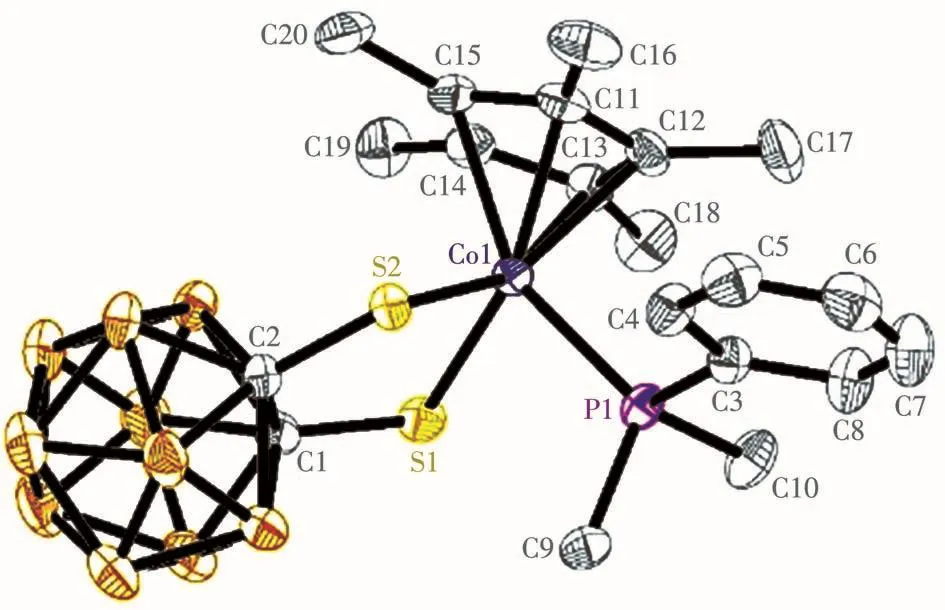
图2 化合物2的椭球几率为30%的分子结构图Fig.2 Mo1ecu1ar structure of compound 2 with therma1 e11ipsoids at 30% probabi1ity 1eve1
化合物3是半夹芯16电子碳硼烷化合物Cp*CoS2C2B10H10和三甲基膦发生加成反应的产物,其分子结构如图3所示。3的碳谱、硼谱、质谱、元素分析和红外光谱等表征数据与3的结构也都相符合。我们在用柱层析方法提纯3时,发现产物3在色谱柱子里会不断分解为原料Cp*CoS2C2B10H10和三甲基膦,这说明硅胶对3的分解有催化作用。这种加成产物在用柱层析进行提纯时会慢慢分解的情况在我们以前的工作中也遇到过[17]。1和2在用柱层析进行提纯时不会分解。
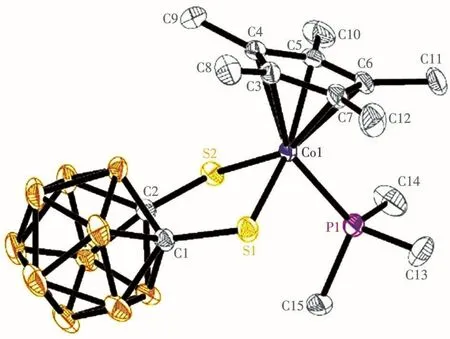
图3 化合物3的椭球几率为30%的分子结构图Fig.3 Mo1ecu1ar structure of compound 3 with therma1 e11ipsoids at 30% probabi1ity 1eve1
与半夹芯16电子碳硼烷化合物CpCoS2C2B10H10比较,Cp*CoS2C2B10H10辅助配体是Cp*。Cp*环上的5个甲基加大了辅助配体的体积,导致Cp*CoS2C2B10H10的反应性下降。例如CpCoS2C2B10H10与PPh3反应生成产物(CpCoS2C2B10H10)(PPh3),而 Cp*CoS2C2B10H10与PPh3不反应[20]。把PPh3换成体积更小的PPh2Me、PPhMe2和PMe3,反应就能顺利发生。这些实验结果进一步说明了辅助配体体积直接影响反应的发生。同时我们还发现一个有趣的现象,就是把产物1和配体PMe3在二氯甲烷中混合,部分产物1分子中的配体PPh2Me会慢慢被体积更小的配体PMe3取代,转变为产物3;但是把产物3和配体PPh2Me在二氯甲烷中混合,并未发现有1生成。
2.2 紫外可见光谱分析和荧光分析
化合物 1、2和 3在乙腈中(10 µmo1·L−1)的 UV-Vis吸收光谱如图4所示,荧光光谱如图5所示。由UV-Vis光谱可知,化合物1、2和3在乙腈中均出现了2个吸收峰,第一个吸收峰分别位于321、316和321 nm;第二个吸收峰分别位于425、399和407 nm,其中波长位于短波的吸收峰对应化合物中共轭体系的π-π*电子跃迁,长波的吸收峰对应分子内电荷转移的电子跃迁[21]。由荧光光谱可知,化合物1、2和3在乙腈中的最大发射波长位于406 nm左右。
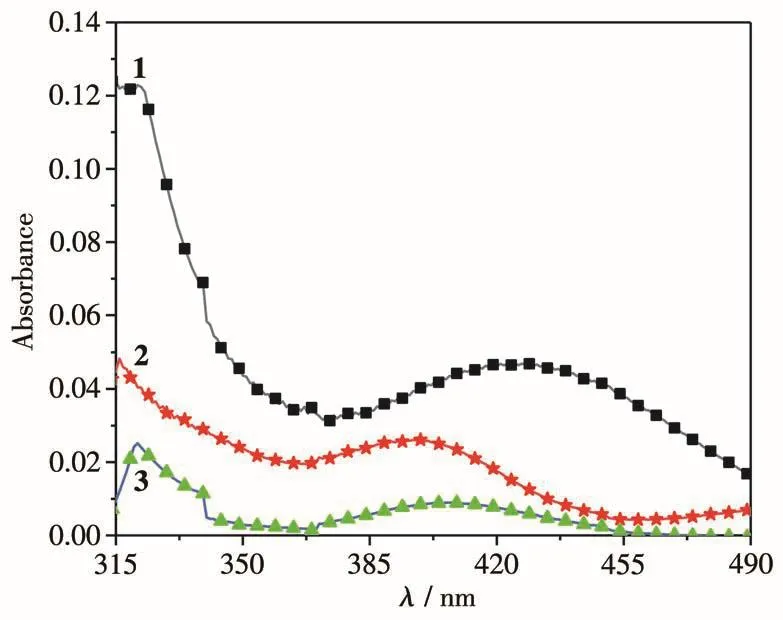
图4 化合物1~3的UV-Vis吸收光谱Fig.4 UV-Vis absorption spectra of compounds 1~3

图5 化合物1~3的荧光光谱Fig.5 F1uorescence spectra of compounds 1~3
