21-羟化酶缺陷症7例临床特点及基因诊断分析
2021-08-10韩宾宾郑瑞芝解一丹袁慧娟
韩宾宾,郑瑞芝,解一丹,袁慧娟
(河南大学人民医院/河南省人民医院 内分泌科,河南 郑州 450003)
先天性肾上腺皮质增生(congenital adrenal hyperplasia,CAH)是类固醇激素合成过程中某种酶缺陷,引起以皮质激素合成受损为特征的一组常染色体隐性遗传性疾病。常见类型包括21-羟化酶、11β-羟化酶、17α-羟化酶等,其中以21-羟化酶缺陷症(21-hydroxylase deficiency,21-OHD)最为常见,占CAH的90%以上[1]。由于CAH临床表型变异较大,基层临床医生对该病认识不足,易出现漏诊或延误诊治,错失治疗良机,且随患者年龄增长可诱发性发育、性心理、性行为等方面的诸多问题,造成其心理与行为偏差,严重影响生活质量。本研究回顾性分析于河南省人民医院就诊的7例21-OHD患者的临床和CYP21A2基因突变特点,以期提高该病的诊治水平。
1 资料与方法
1.1 研究对象回顾性选取2017年11月至2020年6月于河南省人民医院就诊的7例21-OHD患者,其中染色体核型为46,XY 3例,46,XX 4例,年龄3 d~44岁。父母均非近亲婚配,均无家族史。诊断参考《先天性肾上腺皮质增生症21-羟化酶缺陷诊治共识》诊断标准[2],临床均诊断为21-OHD,其中失盐型(salt-wasters,SW)2例、单纯男性化型(simple virilizing,SV)5例。本研究通过了河南省人民医院医学伦理委员会的审查(20170303),患者或家属均签署知情同意书。
1.2 方法
1.2.1生化检查及激素测定方法 全自动生化仪测定血电解质、血糖;血气分析仪测定血气;化学发光法测定黄体生成素(leuteinizing hormone,LH)、卵泡刺激素(follicle stimulating hormone,FSH)、雌二醇(estrogen,E2)、睾酮(testosterone,T)、孕酮(progesterone,P)、雄烯二酮(androstenedione,AND)、硫酸脱氢表雄酮(dehydroepiandrosterone sulfate,DHEA-S)、促肾上腺皮质激素(adrenocorticotropic hormone,ACTH)、皮质醇(cortisol,COR);放射免疫法测定17α-羟孕酮(17α-hydoxy progesterone,17α-OHP)、血浆肾素活性(plasma renin activity,PRA)、血管紧张素Ⅱ(Angiotensin Ⅱ,AngⅡ)、醛固酮(aldosterone,ALD)等。
1.2.2影像学检查 盆腔彩超、肾上腺CT、垂体磁共振成像、左手正位片等。
1.2.3基因检测
1.2.3.1基因组DNA提取 收集患者外周全血标本(EDTA抗凝)备用,采用全血基因组提取试剂盒提取DNA。
1.2.3.2引物设计与合成 根据美国国家生物技术信息中心(National Center of Biotechnology Information,NCBI)中GenBank数据库的CYP21A2基因DNA参考序列,并比较CYP21A1P基因DNA参考序列,使用Primer Premier 5.0软件设计扩增CYP21A2基因全部外显子及旁侧内含子区域(见表1)。

表1 CYP21A2基因10个外显子的扩增引物序列
1.2.3.3PCR扩增与测序 引物各20 pmol,DNA模板0.2 μg,dNTP 10 nmol,MgCl225 nmol,Taq酶 2.5 U。反应条件:95 ℃预变性5 min,95 ℃变性15 s,59 ℃退火30 s,72 ℃延伸40 s,重复35个循环,72 ℃延伸10 min。利用凝胶成像系统观察条带扩增,单一条带者可进行Sanger测序,所有样本均经双向测序验证。若检测到基因突变位点,尚需进一步对受检者父母行靶向变异检测以寻找和明确变异来源。此实验由郑州大学郑州普利莱医学检验所协助完成。
1.2.3.4MLPA检测与分析 采用荷兰MRC-Holland公司的SALSA MLPA kit P050试剂盒按标准操作说明对部分患者及其父母的CYP21A2基因进行检测。
1.2.3.5生物学信息分析 将所有测序结果与文献报道和GenBank数据库中已知的标准参考序列进行比对。
2 结果
2.1 临床特征如表2所示,7例患者中,SW型2例(1男,1女),SV型5例(2男,3女),其中外生殖器异常占85.7%(6/7),儿童期生长加速占71.4%(5/7),皮肤色素沉着占71.4%(5/7),多毛占57.1%(4/7),乳房发育占42.9%(3/7),阴毛早现占28.6%(2/7)。例1(核型为46,XX)生后即发现阴蒂肥大,无失盐表现,查电解质正常,后因体质量不增复诊,查高钾、低钠血症,代谢性酸中毒,确诊为SW型。另有2例女性患者(例4、例7)曾因阴蒂肥大似小阴茎样,分别于4岁、20岁行“外阴整形术”。其中,例4有月经初潮,经期短,经量少,例7为原发性闭经。2例男性SV型患者(例5、例6),均存在幼时生长过快,胡须、喉结过早显现,声音粗或低沉,皮肤粗糙,肌肉发达等男性化表现,但早年未引起重视,以婚后不育就诊。

表2 7例21-OHD患者的临床特征
2.2 实验室检查例1、例2血气分析均示代谢性酸中毒。GnRH激发试验:例3,FSH在0、30、60、90、120 min分别为0.51、2.89、3.43、4.16、4.18 IU·L-1,LH在0、30、60、90、120 min分别为<0.2、1.58、1.54、1.33、1.47 IU·L-1;例4,FSH 25.86 IU·L-1,LH 18.38 IU·L-1。例5、例6患者行精液分析提示无精症。其他内分泌激素结果详见表3。

表3 7例患者激素结果
2.3 影像学检查除1例未查外,余6例肾上腺CT均示双侧肾上腺增粗(见图1)。盆腔彩超示4例女性患者中仅1例未探及,余呈幼稚子宫;2例男性患者(例5、例6)均示双侧睾丸微石症、精囊区囊肿,其中例5曾行“双侧精索静脉高位结扎术+包皮环切术+双侧睾丸切开活检术”,病理未见(院外),例6行“睾丸占位切开取活检术”,病理示“双侧睾丸间质细胞瘤样增生”。已查的5例垂体磁共振成像示例5垂体后异常信号影(考虑Rathke裂隙),余未见异常。3例行左手正位片示2例存在骨龄提前(见图2)。

图1 例6腹部CT(平扫+增强)示双侧肾上腺增生,左侧肾上腺内侧支血管平滑肌脂肪瘤

图2 例3(6岁)左手正位片示骨龄提前(约13岁)
2.4 基因突变分析所有患者均检测出CYP21A2基因变异(见图3),其中例1同时存在CYP21A2基因外显子1~7杂合缺失。共检出7种突变类型(见表4),包括2种剪接突变(c.292+1G>A、c.293-13C/A>G);2种错义突变(c.518T>A、c.1069C>T);2种缺失突变(c.740delA、外显子1~7大片段缺失);1种缺失插入突变(c.1451_1452delGGinsC)。4例行家系成员基因筛查,结果详见表4。

表4 7例21-OHD患者CYP21A2基因的测序结果
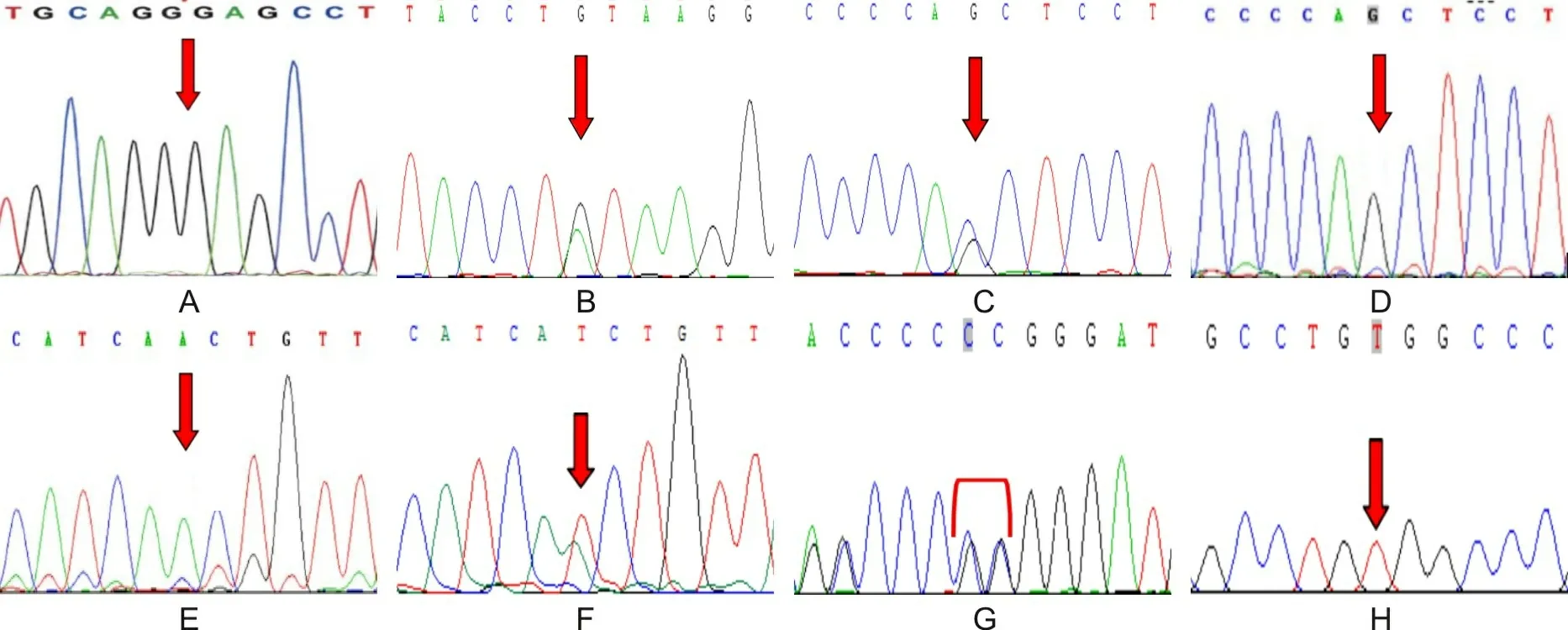
A为例1携带c.740delA纯合变异;B为例2携带c.292+1G>A杂合变异;C为例2携带c.293-13C/A>G杂合变异;D为例5携带c.293-13C>G纯合变异;E为例3携带c.518T>A纯合变异;F为例4携带c.518T>A杂合变异;G为例4携带c.1451_1452delGGinsC杂合变异;H为例6携带c.1069C>T纯合变异。
3 讨论
21-OHD是由编码21-羟化酶的CYP21A2基因突变导致的一种常染色体隐性遗传病,为CAH的最常见类型。该基因突变致酶活性丧失或降低,使通路中孕酮、17-羟孕酮不能正常转化为11-脱氧皮质酮、11-脱氧皮质醇,致皮质醇、醛固酮合成减少,ACTH负反馈分泌增加,肾上腺皮质增生,同时造成上述前体物质大量堆积并向雄性激素转化。CAH主要临床特征为不同程度的肾上腺皮质功能减退、性分化与发育异常、伴或不伴体液平衡紊乱。根据国外流行病学研究显示,典型21-OHD活产儿的发病率为0.07‰~0.1‰,非经典型性的发病率则更高[3-4]。
根据酶缺陷程度的不同,21-OHD可分为经典型和非经典型(nonclassical,NC),前者又分为SW型和SV型[5-6]。SW型,多见于新生儿,常于出生后1~4周出现呕吐、腹泻、喂养困难、脱水、体质量不增、低血糖等非特异性症状就诊,女婴因外生殖器畸形易于早期识别,男婴则不易识别,常因低钠血症、高钾血症、代谢性酸中毒、自发性低血压、休克等肾上腺危象而危及生命。本研究中,例1(女婴)生后即发现阴蒂肥大,无失盐表现,例2(男婴)存在喂养困难、脱水貌、重度营养不良、生长发育落后等失盐表现,随诊中2例均存在高钾、低钠血症,代谢性酸中毒,确诊为SW型。SV型,除无失盐表现、电解质紊乱外,余同失盐型。本研究中,3例女性SV型患者中有2例出现性早熟(例3为外周性,例4为中枢性),另1例女性患者(例7),尽管20岁行外阴整形术,因未明确诊断,术后未能及时补充糖皮质激素,于40岁确诊,至今未婚未育,伴有严重的心理障碍。2例男性SV型患者(例5、例6),均因“婚后不育”就诊,分别于32、33岁确诊。非经典型,症状隐匿,女性多于男性,前者往往以多毛、痤疮、脱发、月经异常、多囊卵巢、不孕症等原因就诊,后者可无症状或仅表现为不育。对于症状不典型的SV型或NC型患者,应尽早行基因检测明确诊断。
CYP21A2基因位于6p21.3,由494个氨基酸组成,与无功能的假基因CYP21A1P高度同源(二者相距30 Kb),与编码补体的C4A及C4B基因相毗邻[7-8]。真假基因间的重排是引起21-OHD致病突变的主要分子病理基础[1,9]。迄今为止,已报道的CYP21A2基因突变类型将近300种[8,10]。众多研究已证实,CYP21A2基因突变谱和热点突变在不同群体之间存在种族差异[11-13]。在55例21-OHD的印度患者中,53例检出双等位基因突变(14例纯合突变,39例复合杂合突变),其中c.293-13C/A>G(20%)突变最为常见,其次为c.1069C>T(14.5%)、c.518T>A(12.7%)和8-bpdel(12.7%)[12]。Wang等[13]对230例中国21-OHD患者(142例SW型、60例SV型、28例待分型)进行CYP21A2基因变异分析,发现最常见的变异位点是c.293-13C/A>G(35%),其次为c.518T>A(14.3%)、c.1069C>T(5.9%),与本研究结果相符。值得一提的是,大片段的缺失/转化被描述为一些地区国家检测到的最常见突变。Chi等[11]对一项来自204个家庭的212名21-OHD越南患者的队列研究显示,缺失突变(占34.48%)为CYP21A2最常见的基因突变类型,其次是c.293-13C/A>G(28.57%)、c.1069C>T(15.51%)和c.518T>A(10.59%)。同样,缺失突变在中国香港、伊拉克、伊朗、德国、奥地利等多个国家的研究中也最常见[14-16]。日本学者Koyama等[17]曾在1例SW型患者中发现c.740delA突变,并预测其会破坏正常阅读框,致P450多肽血红素结合区之前mRNA过早终止,最终导致酶活性完全丧失,本研究中也有此突变位点的报告(即例1的c.740delA纯和突变、外显子1~7杂合缺失)。因此,对于患儿基因突变阳性而父母阴性的病例,应需进一步行MLPA基因检测,以明确有无大片段基因缺失的可能。此外,c.292+1G>A突变位于内含子2经典剪切位置区域,c.1451_1452delGGinsC位于10号外显子,两者均为罕见突变,最初分别被Lee等[18]和Wedell等[19]所报道,预测与SW型21-OHD有关。
21-OHD治疗目标在于激素替代,抑制高雄激素血症,即模拟最佳生长曲线,促进第二性征发育,维持正常生殖功能和预防代谢健康问题(如高血压、糖尿病、胰岛素抵抗、血脂异常、心脑血管疾病等)。其治疗方案的选择有赖于疾病分型、年龄以及有无应激反应、合并症、并发症等。糖皮质激素是此病治疗的关键,部分尚需联合盐皮质激素、性激素替代和促生长药物等综合支持治疗。值得注意的是,对于骨骺尚未闭合的患儿,宜应用氢化可的松,不宜用强的松或地塞米松治疗,以免促进骨骺闭合,影响最终身高。
总之,本研究对象临床表型均符合21-OHD,均存在CYP21A2基因突变,且以纯合突变或复合杂合突变为主,CYP21A2基因可能为21-OHD的致病基因。鉴于21-OHD临床异质性较大,尤其对于非典型患者更容易漏诊或延误诊治,联合基因检测至关重要,有助于该病的早期诊治及改善预后。
