TYMP基因新杂合突变致早发型线粒体神经胃肠型脑肌病1例报告并文献复习
2021-08-05周亚光瞿千千刘海燕崔文豪吕海东
周亚光, 瞿千千, 刘海燕, 崔文豪, 陈 萍, 吕海东
线粒体神经胃肠型脑肌病(mitochondrial neurogastrointestinal encephalomyopathy,MNGIE)是一种罕见的常染色体隐性遗传的多系统疾病,由于核基因TYMP 突变导致胸苷磷酸化酶(thymidine phosphorylase,TP)缺陷所致[1]。TP 的活性降低或缺失,可导致脱氧胸苷和脱氧尿嘧啶核苷在组织中的蓄积,引起线粒体功能的异常,从而导致患者出现多系统受累的临床症状。MNGIE典型的临床症状包括严重的胃肠动力障碍、脑白质病变、眼外肌麻痹、周围神经病变和肌肉萎缩[2]。自2012年许二赫等首次在国内报道了该病以来[3],目前国内约有13例MNGIE的相关报道[4~8]。本文报告1例经肌肉病理与基因检测共同确诊的MNGIE患者,与以往文献报道不同的是本例患者的脑影像不仅有脑白质病变,而且有双侧基底节区和小脑齿状核病变。基因检测发现TYMP基因在7号和10号外显子区域分别有c.914T>C和c.1319T>C两处新发的错义突变,扩展了TYMP基因的变异谱系。
1 资料和方法
1.1 临床特点 患者,男,60岁,农民。以“消瘦、肢体无力35 y,伴双侧眼球活动障碍30 y”为主诉,于2019年3月29日就诊我院。患者自幼有慢性胃肠道病史,间断出现恶心、纳差。25岁时因体型消瘦,肢体力量不如同龄人,不能做重体力劳动。30岁出现双侧眼睑下垂,眼球活动差。曾经以“重症肌无力”口服溴吡斯的明片治疗,症状无改善。45岁以后逐渐丧失体力劳动能力,仅能做一般家务劳动。在当地医院做头部MRI报告双侧大脑半球白质脱髓鞘,以“脑梗死”治疗无好转。近2 y患者消瘦更加明显,并出现反应迟钝,记忆力下降,复查头部MRI提示双侧大脑白质、基底节区、小脑和脑干多发异常信号,为进一步诊治收住院。查体:体型消瘦,身高160 cm,体重46 kg。记忆力、计算力等高级神经功能活动差,MMSE评分23分。双侧眼睑下垂遮盖瞳孔大于1/2,双眼球居中固定,无明显复视,咀嚼和吞咽功能正常。颈伸颈屈肌力均正常,四肢肌张力低,上下肢腱反射均消失,肢体近端肌力4级,远端肌力5级。四肢近远端均有轻度肌肉萎缩,深浅感觉正常。家族史:父母身体健康,非近亲结婚,患者子妹3人,2个姐妹均正常。
1.2 方法 (1)神经电生理:采用日本光电MEB-9200K肌电诱发电位仪,常规进行同心圆针电极肌电图和感觉运动传导速度测定。并行双下肢F波,H反射检测。(2)磁共振:采用GE 1.5T核磁共振对患者脑部做轴位扫描,扫描序列包括T1WI、T2WI及FLAIR序列。(3)肌肉病理:患者在局麻下开放性活检取材,选取右侧股四头肌为活检部位。肌肉标本经液氮固定,恒冷冰冻切片,分别进行HE、MGT、ORO、PAS、ATPase、NADH染色,光镜下观察。(4)基因检测:在患者及其家属知情同意情况下,采取静脉血4 ml,提取基因组DNA,进行全基因组外显子单基因遗传病筛查,由金准基因检验中心进行检测。
2 结 果
2.1 神经电生理 针极肌电图提示左侧股四头肌呈神经源性损害,余被检肌均未见异常;左正中运动神经、右胫运动神经传导速度轻度减慢。
2.2 实验室检查 血常规、血糖、肝肾功能和甲状腺功能均正常,CK 173 U/L,LDH 182 U/L,血乳酸2.73 mmol/L,运动后30 min血乳酸为5.9 mmol/L。
2.3 头部MRI检查 2006年头部MRI提示双侧侧脑室周围、额顶叶皮质下白质对称性、斑片状高信号(见图1A~C);2019年图标MRI报告双侧小脑齿状核、丘脑基底节区、侧脑室周围、额顶叶皮质下及深部白质,均可见弥漫对称性大片状异常高信号,病变范围较前明显扩大(见图1D~F)。

图1 患者头部MRI影像。Flair像:A、B:提示双侧侧脑室体旁和前后角周围异常高信号;C:小脑影像正常(A~C为2006年脑影像);D、E:可见双侧侧脑室体旁和前后角周围大片异常高信号,病变累及双侧丘脑基底节;F:可见小脑齿状核异常高信号(D~F为2019年脑影像)
2.4 肌肉病理检查 经患者同意,行右侧股四头肌活检,病理可见肌纤维大小轻度不等,可见散在有边缘呈嗜碱性的肌纤维,无明显变性坏死肌纤维,亦无炎性细胞浸润;改良Gomori三色可见典型的破碎红边纤维(RRF),未见明显镶边空泡(RV);ORO染色可见部分肌纤维胞浆内有红染的脂肪颗粒增多;ATP酶染色可见Ⅰ、Ⅱ型肌纤维分布异常,Ⅰ型纤维占优势,可见群组化现象(见图2)。
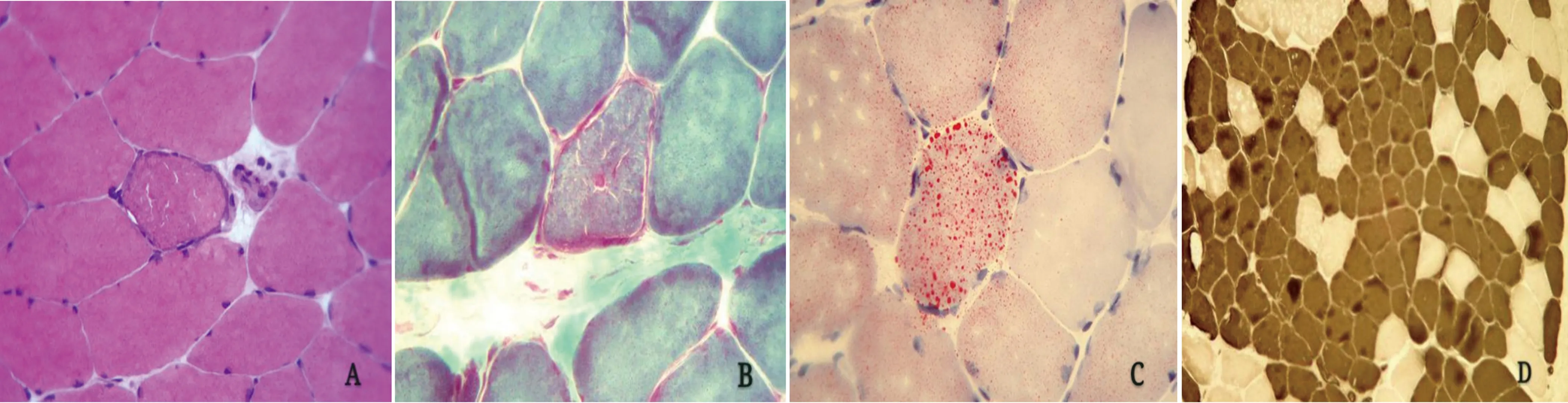
图2 患者肌肉病理图片。A:肌纤维大小轻度不等,可见一个边缘呈嗜碱性的肌纤维 (HE染色,×400);B:改良Gomori三色可见典型的破碎红边肌纤维(RRF)(MGT染色,×400);C:可见少数肌纤维内脂肪颗粒明显增多(ORO染色,×400);D:ATP酶染色可见Ⅰ、Ⅱ型肌纤维分布异常,有群组化现象(ATP酶染色4.3,×100)
2.5 基因检测 发现TYMP基因上有两处新发的错义突变,分别为7号外显子区域的c.914T>C(胸腺嘧啶>胞嘧啶),可导致氨基酸改变p.(leu305Pro),该变异会使所编码蛋白质的第305位氨基酸Leu变为Pro。 另外一个变异发生在10号外显子区域的c.1319T>C(胸腺嘧啶>胞嘧啶),可导致氨基酸改变p.(Val440Ala),该变异会使所编码蛋白质的第440位氨基酸Val变为Ala(见图3)。

图3 TYMP基因Sanger测序图。A:在7号外显子上存在c.914T>C变异;B:在10号外显子上存在c.1319T>C变异
3 讨 论
MNGIE是一种罕见的遗传性线粒体疾病,亦是最早发现的由核基因突变引起的线粒体疾病[9]。MNGIE可累及人体多个脏器,包括胃肠道、骨骼肌、大脑、周围神经和肝脏等,临床表现具有很大的异质性。MNGIE患者从婴儿期到中年均可有发病,但好发于儿童和青少年,平均发病年龄为18岁[10],患者病情随着年龄的增长逐渐进展。MNGIE首发症状多为胃肠道症状占45%~67%,眼外肌麻痹占13%~26%,周围神经病变占8%~13%,以恶病质起病占4%~10%,听力障碍占4%~10%[3]。本例患者在青少年时期以胃肠道症状起病,25岁后逐渐出现肢体无力症状,30岁出现双侧眼睑下垂和眼球活动障碍,45岁以后逐渐丧失劳动能力,也说明MNGIE好发于青少年,且随者患者年龄增长临床症状有逐渐加重的趋势。有文献报道MNGIE的表型,根据发病年龄不同而分为“早发型”和“晚发型”,认为小于40岁发病者为早发型,大于40岁发病者为晚发型[11]。本例患者在青少年时期以胃肠道症状起病,25岁后逐渐出现肢体无力和双侧眼球活动障碍,应当属于早发型MNGIE。MNGIE患者的肢体无力的特点为不能耐受运动和易疲劳,这与其它类型的线粒体肌病相同。不同的是本例患者在发病30多年后,逐渐出现记忆力下降、反应迟钝等高级神经功能障碍,临床表现由原来的肌无力和胃肠道症状,逐渐转变为高级神经功能障碍为主的临床症状。脑白质病变是MNGIE患者的特征之一,而且很容易被误诊为脑血管病和其它脑白质病变。本例患者在45岁时做头部MRI发现脑白质病变,曾经误以脑梗死进行治疗。此次就诊复查头部MRI可见脑白质病变范围较15 y前明显扩大,而且在双侧丘脑、基底节和小脑齿状核,亦发现有异常高信号,此在以往文献中少有报道。我们认为这些多发对称性的脑白质病变和基底节核团病变,是导致患者出现高级神经功能损害症状的病理基础。
骨骼肌活检对MNGIE患者的诊断具有重要的临床意义,其特征性病理改变是在肌组织内可见破碎红边纤维(RRF)[12]。在本例患者肌肉病理上,我们不仅发现有破碎红边纤维,而且ORO染色可见部分肌纤维内脂质明显增多,表明MNGIE患者亦存在有脂质代谢异常。ATP酶染色可见两型肌纤维分布明显异常,有群组化现象,提示有神经源性损害,这也从病理上间接证实了MNGIE患者可以合并周围神经损害的观点。
TYMP基因是导致MNGIE的主要致病基因[10,13],其位于22号染色体长臂的13区32带[14],它所编码的胸苷磷酸化酶(TP)是脱氧胸苷和脱氧尿苷代谢所必须的酶。MNGIE患者的TP活性下降或缺失,使血液和组织中的脱氧胸苷和脱氧尿苷毒性蓄积,导致线粒体内脱氧核糖核酸供应失衡,影响线粒体DNA的复制,最终导致线粒体功能受损[15,16]。 TYMP基因含有10个外显子,1号外显子为调节区,2-10外显子为编码区[17]。目前人类基因突变数据库已报告TYMP基因有97种不同的变异,这些变异位点分别被定位到外显子或内含子区域[18]。已经报道的变异类型包括错义、复制、删除、单碱基插入和内含子的剪接等[19]。本例患者在TYMP基因上发现两处新发的错义突变,分别为2号外显子区域的c.914T>C和10号外显子区域的c.1319T>C,两者均可导致氨基酸的改变。这两个变异位点在人类基因数据库和HGMDpro数据库中均未见有报道,认为是TYMP基因新发的突变位点。
有关MNGIE的诊断标准,目前多采用Hirano[20]等提出的诊断标准:(1)胃肠动力障碍;(2)眼睑下垂和(或)眼外肌麻痹;(3)周围神经病变;(4)肌肉活检发现破碎红边纤维。实验室检查包括:(1)基因检测发现TYMP基因突变;(2)头部MRI提示白质脑病;(3)检测白细胞中TP的活性,血浆或尿中脱氧胸苷(dThd)和(或)脱氧尿苷(dUrd)水平。本例患者具有典型的胃肠道症状、脑白质病变、骨骼肌无力、眼睑下垂及眼外肌麻痹。同时肌肉活检可见典型的破碎红边纤维(RRF),二代基因测序证实有TYMP基因突变,因此本病例确诊为MNGIE无疑。
目前MNGIE尚没有特效疗法,主要是对症支持治疗。可给予辅酶Q10、ATP、维生素E、维生素B2、烟酸等药物,对改善线粒体的功能有一定的作用[9]。近年来有文献报道造血干细胞移植(HSCT)能够恢复一定的TP酶活性,降低血浆中胸苷和脱氧尿苷的浓度,使其接近正常范围[21]。但其逆转或明显改善临床症状的能力尚未得到证实[22]。红细胞包裹胸苷磷酸化酶(EE-TP)是一种新兴的治疗MNGIE的酶替代疗法,其优点是侵袭性小且安全性高,但其疗效不如造血干细胞移植[23]。
综上所述,线粒体神经胃肠型脑肌病早期以胃肠道、眼外肌、骨骼肌症状为主,随着年龄的增长临床症状逐渐加重,后期可出现高级神经功能活动障碍。头部MRI影像不仅有脑白质病变,还可出现双侧小脑齿状核和丘脑基底节区病变。肌肉活检发现破碎红边纤维和基因检测发现TYMP突变是诊断MNGIE的重要依据,我们检测到c.914T>C和c.1319T>C两个新的变异位点,扩展了TYMP基因的变异谱系。
