GCH1基因新的移码突变引起的多巴反应性肌张力障碍1例报告和简要回顾
2021-08-05胡建萍张廷华
胡建萍, 张廷华, 郭 佳
多巴反应性肌张力障碍(Dopa-responsive dystonia,DRD)[1]是1976年由Masaya Segawa首次报道,又称Segawa病,其临床特征包括肌张力障碍、步态障碍、僵硬、轻度帕金森病、日间波动和对左旋多巴的剧烈反应等。患者通常对左旋多巴反应良好,症状明显改善,甚至消失。多巴反应性肌张力障碍(DRD)是一种可治疗的遗传性疾病,因此识别它是非常重要的。本研究拟通过报道1例DRD GCH1基因新的移码突变,以提高临床对DRD诊断及预后的认识,以免误诊。
1 病例资料
患者,女,47岁,因“进行性行走不稳30 y余。”入院。患者于入院前30余年无明显诱因出现行走不稳,走路右足跟不着地,右手摆动少,渐出现右侧肢体发僵,走路不自然,易跌倒,逐渐累积左侧肢体,自觉四肢僵硬,并出现四肢抖动,行走时脚后跟不能着地,上述症状有晨轻暮重现象,当时未在意,后上述症状呈缓慢加重趋势,出现行走困难,需要外界帮助。随就诊我院收住入院。入院查体:神情,精神欠佳,问答切题,查体合作。颅神经(-)。颈软,四肢静止性震颤,肌张力增高,双侧上下肢肌力3级,双侧病理征阴性。深浅感觉(-)。其父亲有类似较轻表现,母亲无类似症状。入院后完善头部+脊柱MR:(1)头部MR平扫未见明显异常,双侧上颌窦炎;(2)全脊柱轻度退行性变,颈椎曲度变直;(3)颈5/6椎间盘突出,相应水平脊髓略受压,腰4/5、腰5/骶1椎间盘轻度膨出;(4)骶2椎体水平骶管内小囊肿;(5)腰椎水平背部皮下软组织少量渗出,所示少量盆腔积液,结合临床。血常规生化、传染病、垂体功能检查均未见明显异常。甲状腺功能检查甲状腺球蛋白抗体296.90 U/ml(参考值:0.00~60.00 U/ml),甲状腺过氧化物酶抗体>1300 U/ml(参考值:0.00~60.00 U/ml),余指标正常范围。甲状腺B超检查:甲状腺未见明显异常。眼科检查未见K-F环。内分泌科会诊考虑桥本甲状腺炎,建议6 m后复查甲状腺功能。患者病史时间长,以锥体外系症状为主,有晨轻暮重趋势。诊断多考虑多巴反应性肌张力障碍。给予小剂量美多芭口服,服药后第2天症状改善,最终给予美多巴62.5 mg/d,3次/d,症状改善理想。并完善基因检查,取患者及父母静脉血3 ml送全外显子组测序检测显示患者(见图1)GCH1基因c.192_c.193insC(p.E65Rfs*6)杂合突变,该突变导致GCH1基因所编码的蛋白发生移码突变。在患者的DRD基因报告中第14号染色体长臂的GCHl基因第1号外显子检测到一移码突变c.192_c.193insC(p.E65Rfs*6),此突变将导致开放阅读框后移,自65位氨基酸谷氨酸突变为精氨酸,向后翻译到第6位时遇到终止密码子,蛋白质翻译终止,预计产生截断蛋白或导致无义介导的mRNA降解(nonsense-mediated mRNA decay,NMD)。该变异未见与疾病发生相关报道,数据库HGMD和LOVD未见收录。ACMG致病性分析。该变异在1000Genomes、ExAc和gnomAD人群频率数据库未收录,符合致病证据PM2;该变异属于移码变异,功能丧失是GCH1基因的致病机制,符合致病证据PVS1。综上所述,根据ACMG标准该变异判定为可能致病变异。为进一步明确突变来源,又继续检测了先证者的父母,其父亲有类似较轻的症状,基因检测显示先证者父亲c.192_c.193insC(p.E65Rfs*6)移码突变(见图2),而其母亲为正常野生型(见图3)。基因检测为常染色体显性遗传,该变异先证者为杂合子,符合常染色体显性遗传(AD)的疾病发病机制,与患者表型存在相关性;先证者及其家系成员表型及基因型符合共分离。结合患者病史、查体、基因检测及治疗反应最终该患者诊断为DRD。
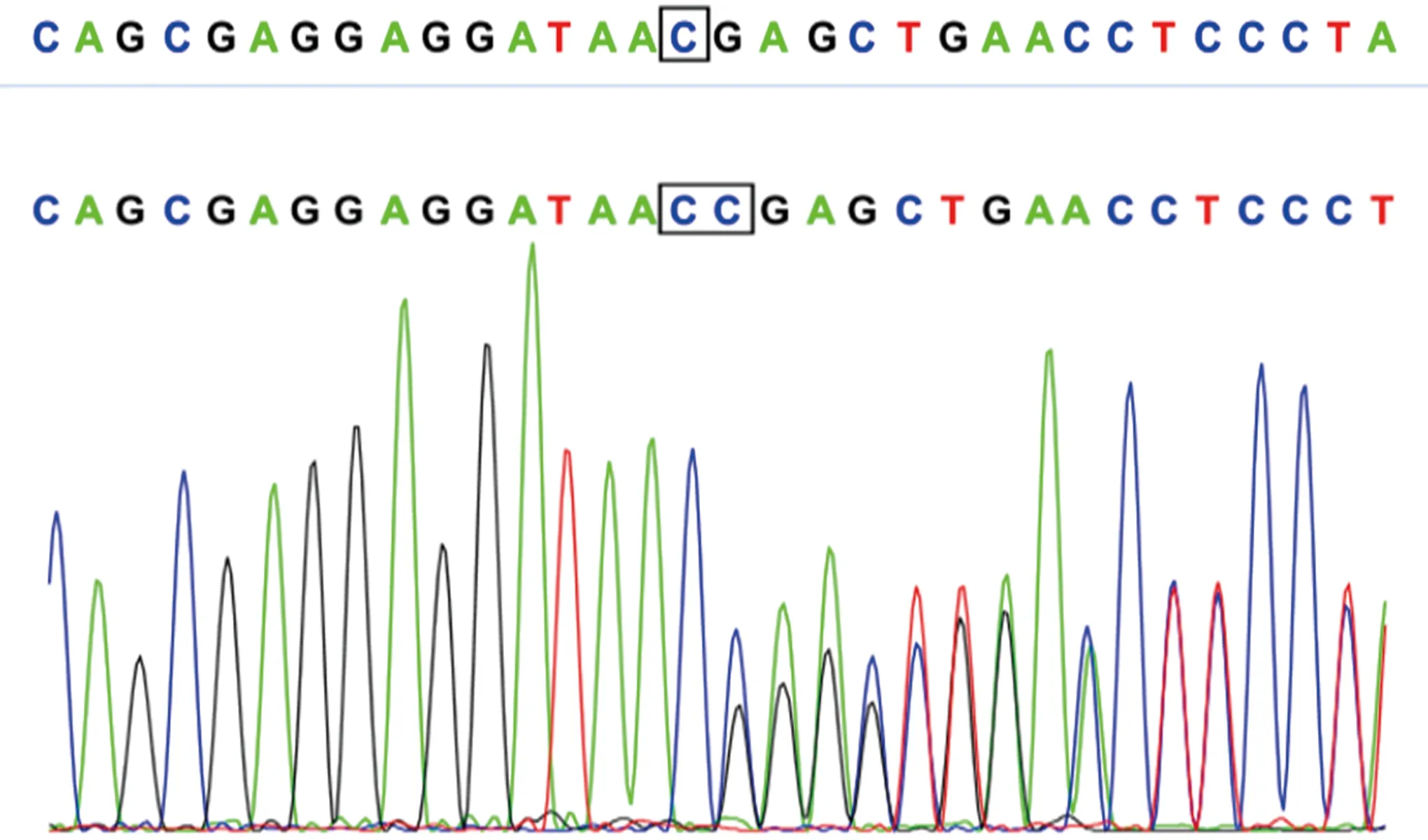
图1 先证者,患者,杂合cGCH1:c.192(exon1)_c.193(exon1)insC

图2 其父,轻微表型,杂合cGCH1:c.192(exon1)_c.193(exon1)insC

图3 其母,正常,野生型
2 讨 论
多巴反应性肌张力障碍(DRD)是一种比较复杂的遗传性疾病,有常染色体显性遗传和常染色体隐性遗传,最常见的是常染色体显性遗传[2]。常染色体显性遗传中最常见的病因是编码GTP环水解酶(GCH1)的基因突变,常染色体隐性遗传中最常见的病因是编码酪氨酸羟化酶(TH)的基因突变[3]。DRD家系中发现的第1个致病基因是位于14号染色体(14q22.1-14q22.2)的GCH1基因[4]。患者的GCH1基因突变来源于父母[5]。在DRD中发生点突变大约54%,其中错义突变占47%,内含子剪接突变占44%,无义突变占6%,移码突变占3%;此外,约5%患者发生基因缺失[6]。
本例先证者基因检测显示GCH1的c.192(exon1)_c.193(exon1)insC为可疑致病移码突变。因该突变导致相应蛋白编码错误,致使蛋白失去相应功能,继而发病。为进一步明确突变来源,又继续检测了先证者的父母,其父亲有类似较轻的症状,基因检测显示先证者父亲c.192_c.193insC(p.E65Rfs*6)移码突变,基因检测为常染色体显性遗传,该变异先证者为杂合子,符合常染色体显性遗传(AD)的疾病发病机制,与患者表型存在相关性;先证者及其家系成员表型及基因型符合共分离。先证者之父携带该基因突变,故突变来源在本谱系其父。
DRD是由于纹状体通路上酪氨酸羟化酶缺乏,从而影响多巴胺合成减少[7]。DRD发病较早,一般为1~12岁,平均6岁左右,个别可晚至50~60岁间发病,发病率女性多于男性,男∶女=1∶4[8]。常见的症状包括步态异常、肌肉僵直、运动问题等,非典型症状包括流口水、吞咽问题、肢体抽搐和小脑功能异常等[9]。儿童期肌张力障碍发病从一侧下肢开始,表现步态怪异、下肢僵硬、马蹄内翻足等。有时表现为学走路较迟,容易摔跤,随病情的发展,肌张力异常影响到其他肢体,甚至头颈部及身体中轴,出现痉挛性斜颈,扭转痉挛等[10]。患儿可有肢体震颤,肌强直及病理征阳性,言语及智能一般不受累。成人以四肢不自主震颤、僵硬感等类似帕金森综合征表现比较多见。大约有75%的DRD患者症状有昼夜波动性,晨起或休息后可减轻甚而消失,下午或劳累后症状加重,该病需与肝豆状核变性、脑性瘫痪、痉挛性截瘫、少年型帕金森病等相鉴别[11]。本例先证者发病年轻,病情进行性加重,早起生活能自理未予重视,表现为步态异常,肢体不自主震颤、僵硬,类似帕金森综合征表现,与典型DRD临床表现基本相符。
使用小剂量左旋多巴可显著改善症状是DRD的典型特点,即便数十年未经治疗的患者仍有很好的药物反应性[12]。左旋多巴的初始建议剂量成年人50 mg,qd,且在妊娠期间继续治疗多数无不良影响[6]。本例先证者最终在服用美多巴(左旋多巴/苄丝肼)62.5 mg,tid后症状明显改善。
总之,DRD临床症状丰富多样,易与其他疾病混淆,导致治疗延迟。本研究通过本例家系的报道及GCH1基因突变新位点的发现,并扩充了GCH1基因突变谱。结合既往文献,总结了DRD的临床特点及预后,有利于提高临床上对DRD的识别。
