Stem cell therapy: A paradigm shift in breast cancer treatment
2021-07-30SabihaKhanMoushumiSuryavanshiJasamritKaurDebadattaNayakAnilKhuranaRajKumarManchandaChanderdeepTandonSimranTandon
Sabiha Khan, Moushumi Suryavanshi, Jasamrit Kaur, Debadatta Nayak, Anil Khurana, Raj Kumar Manchanda,Chanderdeep Tandon, Simran Tandon
Sabiha Khan, Simran Tandon, Amity Institute of Molecular Medicine and Stem Cell Research,Amity University, Noida 201313, Uttar Pradesh, India
Moushumi Suryavanshi, Department of Pathology, Rajiv Gandhi Cancer Institute and Research Centre, New Delhi 110085, India
Jasamrit Kaur, Department of Chemistry, Goswami Ganesh Dutta Sanatan Dharma College,Chandigarh 160030, India
Debadatta Nayak, Anil Khurana, Central Council for Research in Homeopathy, New Delhi 110058, India
Raj Kumar Manchanda, Department of AYUSH, Karol Bagh, New Delhi 110011, India
Chanderdeep Tandon, Amity Institute of Biotechnology, Amity University, Noida 201313, Uttar Pradesh, India
Abstract As per the latest Globocan statistics, the high prevalence rate of breast cancer in low- and middle-income countries has led to it becoming the most common cancer to be diagnosed, hence posing a major public health challenge. As per this data, more than 11.7% of the estimated new cancer cases in 2020 were due to breast cancer. A small but significant subpopulation of cells with self- renewing ability are present in the tumor stroma and have been given the nomenclature of cancer stem cells (CSCs). These cells display a high degree of plasticity owing to their ability to transition from the slowly cycling quiescent phase to the actively proliferating phenotype. This attribute of CSCs allows them to differentiate into various cell types having diverse functions. Breast CSCs have a pivotal role in development, metastasis, treatment resistance and relapse of breast cancers. This review focuses on the pathways regulating breast CSC maintenance and the current strategies that are being explored for directing the development of novel,targeted, therapeutic approaches for limiting and eradicating this aberrant stem cell population.
Key Words: Molecular genomics; Breast cancer stem cells; Tumor microenvironment;Molecular pathways; Targeted therapy; Inhibitors
INTRODUCTION
Breast cancer (BCa) is a complex, heterogeneous disease with distinct cancer subtypes,identified through gene expression profiling. BCa subtypes can also be classified through simpler, biological evaluation of specific surface markers and histopathological features such as size, shape and arrangement of BCa cells[1,2].
The breast is a unique organ in the sense that it develops postnatally and in order to acquire its distinctive morphology and perform specific functions, has a large pool of mammary stem cells (MaSCs). These MaSCs have the potential to give rise to the progenitor cells of the luminal and basal epithelial lineages, which further differentiate to give rise to the various cell types that constitute the breast. The MaSCs are present in a dynamic microenvironment that allows them to respond to the diverse signals of the transforming growth factor beta (TGF-β), Wnt, fibroblast growth factor, Hedgehog(Hh), epidermal growth factor, estrogen and Notch pathways. This cellular communication leads to the characteristic changes in the epithelial phenotypes of the mammary tissue seen during puberty, menstrual cycles and pregnancy, which are repeated many times in the reproductive period in women[3]. This ability to repeatedly respond, divide and differentiate puts these MaSCs at risk for acquiring mutations, which can lead to transformed stem cells, thereby promoting the process of tumorigenesis[4]. Increasing evidence points to the presence of a small subpopulation of cancerous cells, termed as BCa stem cells (BCSCs), which have unlimited renewal capacity and compared to MaSCs possess highly deregulated molecular signaling pathways. BCSCs are known to maintain a highly controlled microenvironment for their growth and survival and are responsible for repopulating and giving rise to the heterogeneous, overly aggressive tumors, indicative of cancer relapse following treatment[5].
In this review we have discussed the current knowledge on major oncogenic pathways regulating BCSCs, their role in disease progression and as possible targets for therapeutic intervention, leading to more promising disease outcomes.
MOLECULAR CLASSIFICATION OF BREAST CANCER
Data derived from signaling pathways and gene and protein expression analysis has helped in the molecular classification of BCa. BCa is classified as estrogen receptor(ER) positive (luminal A and luminal B), which account for 60%-65% of BCa and the essentially ER negative, basal-like and epidermal growth factor receptor 2 (HER2)-positive subtypes. Triple-negative BCa (TNBC) shows absence of ER, progesterone receptor and HER2 based on hormone receptors status and copy number alterations,which leads to deletions or amplifications of large regions of the genome.
The various cancer subtypes are associated with distinct tumor characteristics and clinical outcomes. The luminal type cancers show the most recurrent mutations;however these cancers have the most favorable prognosis as they respond well to hormonal therapy[6,7]. Cancer survival statistics reveal that the prognosis of luminal B subtype, which is of a higher grade, is comparatively poorer compared to luminal A[8]. Amongst the ER negative tumors, basal-like tumors show the worst outcomes, and the genes display frame shift, nonsense and complex mutations[9]. Around 75% of TNBC subtype are also categorized as basal-like subtype based on gene expression profiling. In HER2 type, which accounts for 20%-30% of BCa, co-amplification of genes occurs in the HER2 amplicon and is associated with an aggressive, metastatic disease[10]. The genomic landscape of TNBC reveals wide ranging DNA instability and accounts for the observed heterogeneity due to DNA copy number and transcriptional variability[11].
SIGNALING CUES CONTROLLING BREAST CANCER PROGRESSION
There are various pathways that are involved in cancer cell growth and survival,which include phosphatidylinositol 3-kinase (PI3K)/Akt, HER2, cyclin dependent kinase (CDK), nuclear factor kappa beta (NF-κB) and breast cancer type 1 (BRCA1)[12].
PI3K-enhancing mechanisms have been widely reported in BCa and are linked with endocrine therapy resistance[13]. The mutated or amplifiedPIK3CAgene in ER+ BCa results in increased downstream signaling and carcinogenesis[14,15].
HER2 expression levels are a critical diagnostic marker for breast tumorigenesis,and hence HER2 status is an important determinant before initiating hormone therapy[16,17]. Reports suggest that HER2 overexpression generates a multidrug resistance phenotype actingviathe PI3K/Akt pathway[18].
One important and classical hallmark of cancer is the deregulation of the cell cycle.CDKs are the pivotal drivers of cell cycle, which are positively or negatively controlled by cyclins and CDK inhibitors, respectively[19]. Targeting CDKs using synthetic inhibitors is a promising treatment regimen against cancer[20].
The transcription factor NF-κB plays an important role in various biological processes controlling cell survival, proliferation, inflammation and immunity and has been shown to be constitutively activated in various cancers[21]. Various studies have shown that in ER- compared to ER+ breast tumors, higher expression of activated NFκB were detected[22].
The tumor suppressorBRCA1susceptibility gene is frequently mutated in cancer,leading to widespread chromosome instability and is statistically correlated with a 55%-65% lifetime risk in women developing BCa[23].The loss of BRCA1 is responsible for the formation of aggressive basal-like BCa and expansion of BCSCs by modulating various pathways such as JAK-STAT, Hh, Notch and PI3K[24,25].
The emerging role micro RNAs (miRNA) in regulation of diverse cancers and cancer stem cells (CSCs) points to the importance in understanding this pathway for effective management of BCa. Research studies have shown that miRNAs can act either as tumor suppressors or as oncogenes and impact breast tumorigenesis and progression as they are associated with modulation of genes linked with CSC pathways[26].
CSCS AND TUMORIGENESIS
Tumors have a small subpopulation of undifferentiated cells with self-renewal,stemness and regeneration capability[27]. These cancer cells owing to their distinct similarity with the stem cell population found in normal tissues have been given the nomenclature of CSCs or tumor-initiating cells and make up 0.1%-1.0% of the tumor bulk. The first markers identified for CSCs were CD44, CD24 and later the enzyme aldehyde dehydrogenase (ALDH or ALDH1) was added to the panel, allowing for their isolation using fluorescence-activated cell sorting or immune selection methodologies[28].
The Cancer Stem Cell theory has its basis in data obtained from studies of human acute myeloid leukemia by the research group of Bonnet and Dick who were able to show that the transplantation of only a few leukemia cells had the ability to produce new tumors in mice[29]. This subpopulation of CSCs in the tumor can self-renew and differentiate, giving rise to the non–stem cancer cells that comprise the bulk of the tumor. It has been reported that specifically in BCa, the tumor bulk is derived from BCSCs[30]. These BCSCs are characterized as CD44+/CD24-/Lin-. The origin of CSC is still highly debatable, but various theories have been put forward (Figure 1). The first one states that normal long-lived stem cells become malignant over the course of time as they accumulate genetic mutations. The second theory proposes that mutations in a lineage-committed cell could result in a gain of stemness function[31].
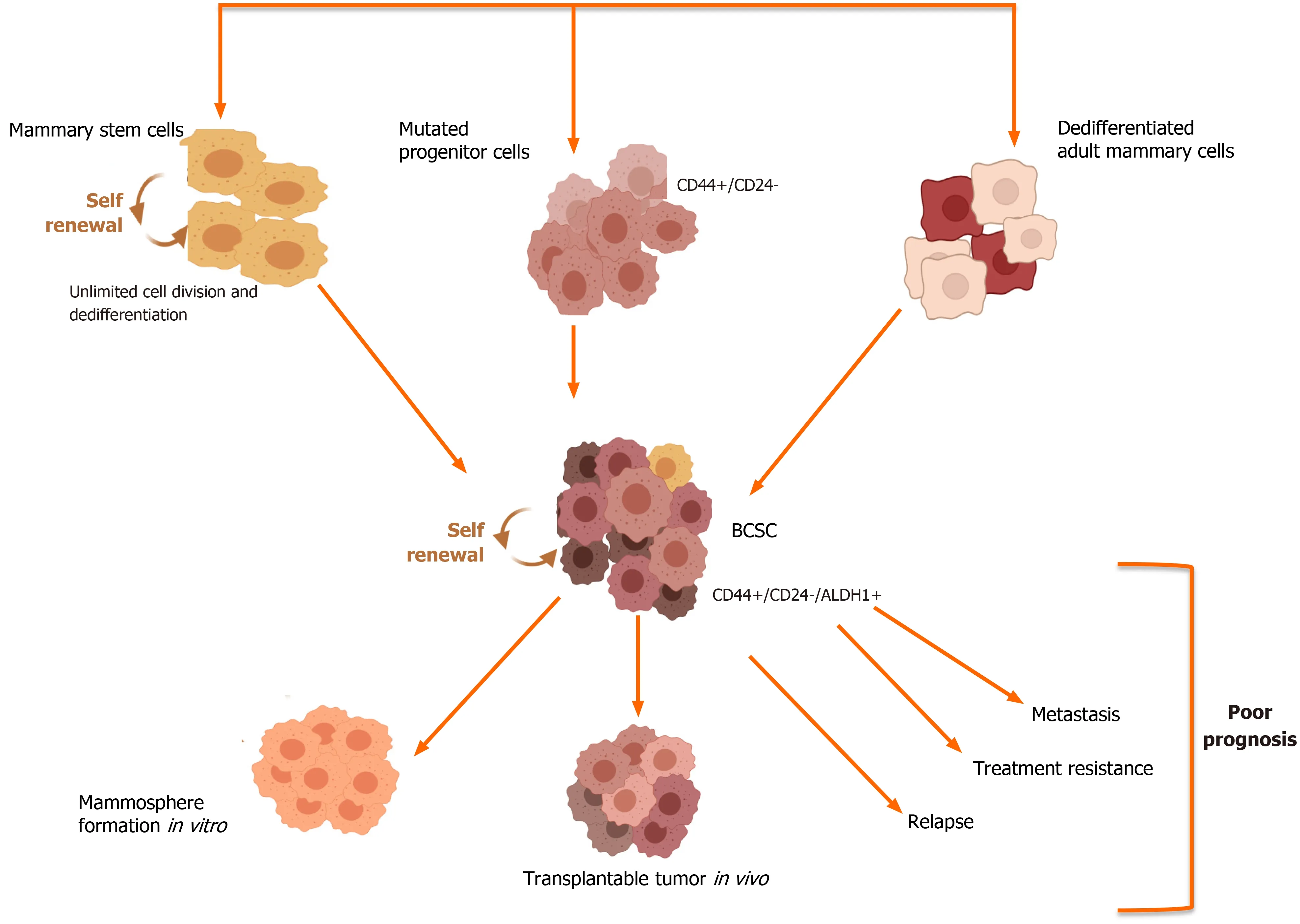
Figure 1 Breast cancer stem cells: Origin and outcome. Breast cancer stem cells (BCSCs) can arise from normal mammary stem cells which have undergone mutations or from mutated progenitor cells or dedifferentiated cells, which comprise the mammary tissue. The subpopulation of BCSCs can be identified based on the altered expression of cell surface markers such as CD44 and CD24 as well as higher activity of the enzyme aldehyde dehydrogenase (ALDH1). These BCSCs when transplanted in SCID/nude mice have the ability to form mammospheres, which are three-dimensional spheres generated through the clonal expansion of single cancer stem cells. These cancerous stem cells have been implicated in the poor outcomes associated with aggressive breast cancer subtypes.
In 2003, the identification of BCSC from the normal MaSCs was shown for the first time by Al-Hajjet al[32]. Several studies have shown that these CSCs are responsible for the poor prognosis seen in cancer, as they impact tumor metastasis, recurrence and resistance to conventional chemotherapy and radiotherapy[33,34].
The importance of research on BCSCs, which has gradually gained grounds as key drivers of tumorigenesis, could be pivotal in understanding the progression as well as prognosis of cancer.
PATHWAYS REGULATING CSCS
Pathways responsible for self- regeneration and survival of BCSCs include Notch,Wnt, Hh, PI3K/Akt/mTOR, TGF-β and HER2[35]. As CSCs have a major role in BCa progression and metastasis, elucidation of key signaling pathways involved will prove instrumental in the development of suitable drugs to target and eliminate CSCs and prevent disease relapse.
The manner in which these pathways influence the stemness phenotypes can be attributed to the aberrant activation of the various effectors in select signaling pathways (Figure 2).
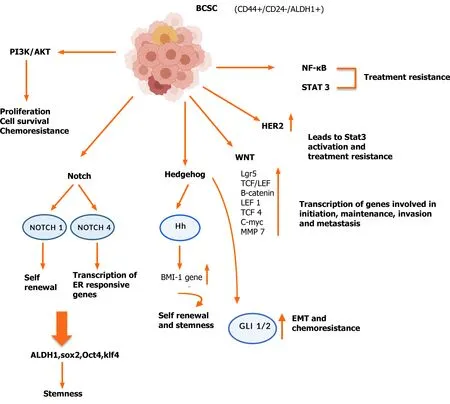
Figure 2 Dysregulated pathways operative in breast cancer stem cells. A number of pathways have been shown to be upregulated in breast cancer stem cells, which are due to mutations, resulting in amplification or loss of key effector genes. The gene products of the aberrant pathways impart a growth and survival advantage and allow these cells to metastasize and become resistant to conventional therapy. BCSC: Breast cancer stem cell; NF-κB: Nuclear factor kappa beta; PI3K: Phosphatidylinositol 3-kinase; ER: Estrogen receptor; EMT: Epithelial mesenchymal transitions; HER2: Epidermal growth factor receptor 2; ALDH1:Aldehyde dehydrogenase; Hh: Hedgehog.
Notch pathway
Notch pathway is a key mediator of numerous cellular and developmental processes as well as a key physiological player in cellular differentiation and self-renewal[36]. In addition, abnormal activation of Notch pathway is linked to epithelial mesenchymal transitions (EMT) and cancer metastasis[37]. High expression of specific Notch receptors and ligands is an indicator of poor prognosis, as this leads to treatment resistance in BCa[38]. Studies have shown that knockdown of the Notch 4 receptor by either genetic manipulation or by drugs can directly impact BCSCs, implying the crucial role of Notch 4 on CSC development[41].
In light of the diverse signaling crosstalk between Notch and several other pathways such as Ras, TGF-β, Hh and Wnt, which is vital for tumor progression[42],targeting Notch alone or in combination with other pathways could represent a promising therapeutic strategy for the effective management of hard-to-treat BCa.
Hh pathway
Hh signaling is a pivotal morphogenic driver regulating cancer initiation, progression and metastasis in specific cancer types and has also been reported to maintain stemness[43]. GLI1 has been shown to regulate CSCs, and studies have shown that estrogen promotes CSC development and EMT through GLI1, which could account for the BCSC-induced endocrine resistance in ER+ BCa[44]. Hh signaling causes mitogenesis of normal MaSCs by the differentiation of mammary epithelial progenitor cells through TP63 transcription factor, which regulates the expression of the various effectors Shh, GLI2 and PTCH1 and generates a positive feedback for mammary progenitor cells[45].A similar picture, although acting in an amplified manner, is also operative in BCSCs. The secretion of Shh by BCSCs has been shown to upregulate cancer-associated fibroblasts, which in turn promotes the proliferation and selfrenewal of the BCSC pool by secreting various factors[46].
These findings reiterate that Hh signaling regulates the stemness of CSCs and offers a novel therapeutic strategy for targeting this pathway for improved overall survival[47].
Wnt signaling
Evidence for the role of Wnt signaling in BCSC regulation is attributed to deregulation of this pathway[49]. The Cancer Gene Atlas through large-scale genome sequencing is a comprehensive, molecularly characterized database of over 11000 patients consisting of over 20000 primary cancer and matched normal samples, spanning 33 cancer types over a 12-year period. The Cancer Gene Atlas has shown the presence of a large number of mutated Wnt genes[50] (Table 1).
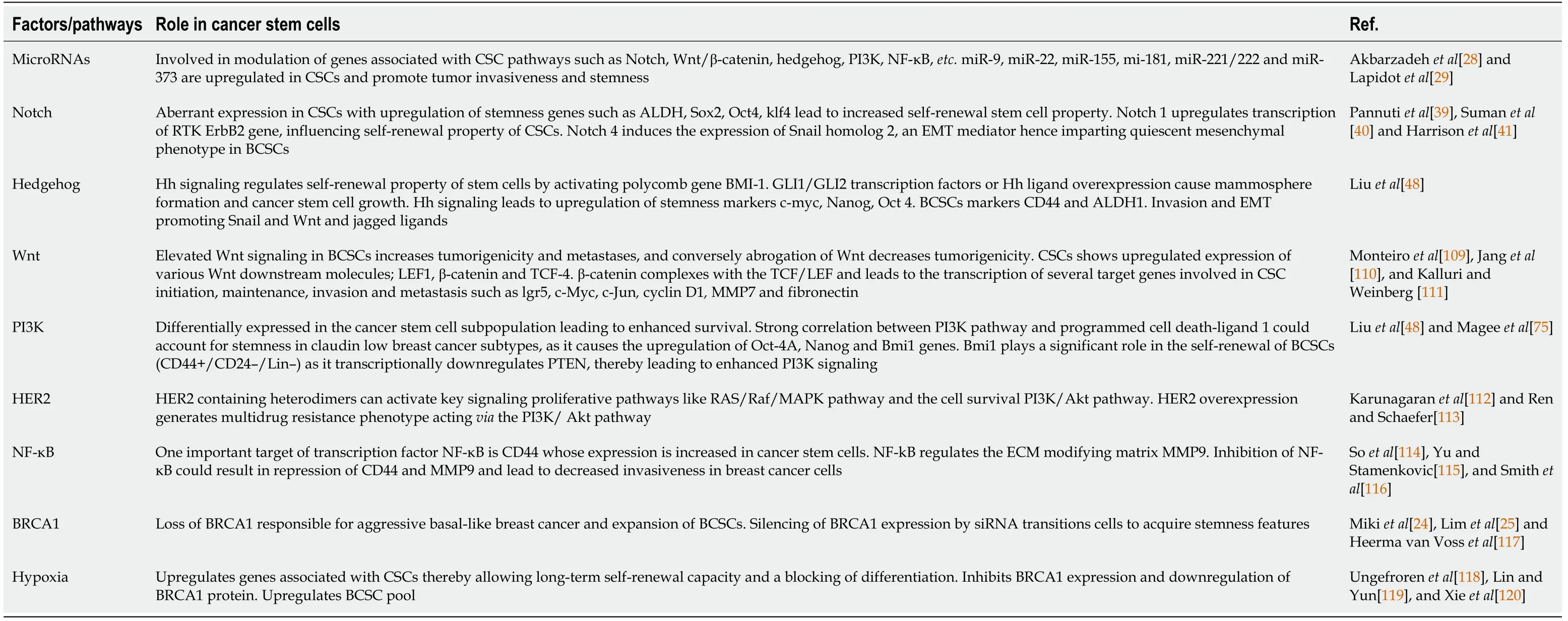
Table 1 Pathways and factors influencing cancer stem cells
BCSC MARKERS
Although no universal markers specifically for BCSCs have been identified, these cells can be recognized based upon the expression of specific surface markers: CD44, CD24 and ALDH1+, which are the most accepted biomarkers for the identification of the BCSC phenotype[34]. Studies in SCID mice which were transplanted with a small number (around 100 cells) of these BCSCs (CD44+/CD24-/Lin-) showed the development of tumors with a similar molecular heterogeneity as the original tumor,validating that CSCs as drivers of cancer recurrence, metastasis and treatment resistance[51]. Clinical data has shown that cells with CD44+CD24-/Lowand ALDH1+expression are more tumorigenic and result in poor survival rates in patients[52].
CD44, a transmembrane glycoprotein that acts as a receptor for various growth factors and ligands present in the extracellular matrix[53], is responsible for maintenance of BCSC population by activating Ras-MAPK and PI3K/AKT pathways[54] and imparts treatment resistance through the activation of p62-associated nuclear factor erythroid2-related factor 2[55]. A direct correlation between CD44 expression and NF-κB activation has been seen to result in radioresistance and poor prognosis.CD24-/Lowconfers treatment resistance, and the ALDH enzyme is responsible for the conversion of retinol to retinoic acid, which is required for the differentiation property of normal and malignant stem cells[56].
The CD44+/CD24-/Lowsurface biomarker expression on BCSC is an indicator of a more invasive, quiescent, basal phenotype, while BCSCs with the ALDH1+epitheliallike phenotype show a proliferative and localized luminal nature[57]. The BCSCs expressing CD44+/CD24-/Lowand ALDH1+display elevated tumorigenic and metastatic activity[58]. BCSCs show a higher expression of an isoform of CD44, CD44v6, which can bind to hepatocyte growth factor, osteopontin and other major cytokines produced by the tumor microenvironment (TME) and aid in cancer progression, cell migration and invasion[59].
Additional markers for BCSCs include CD133, CD49f, CD61 and CD29, which are also expressed on normal MaSCs (Figure 3). The use of these markers along with CD44+/CD24-/Low/ALDH1+can be highly useful for the isolation of BCSCs as well as possible prognostic markers[60].
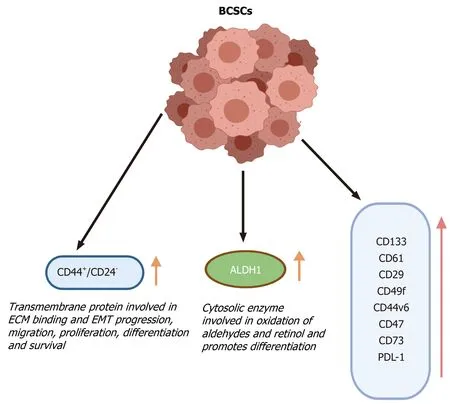
Figure 3 Breast cancer stem cell markers. The breast cancer stem cells can be differentiated from cancer and normal cells based on the increased or altered expression of various cell surface markers. These marker proteins have various functions that are responsible for the development and survival of breast cancer stem cells and can be used to isolate the cancer stem cells from the bulk of the tumor. BCSCs: Breast cancer stem cells; ECM: Extracellular matrix; EMT: Epithelial mesenchymal transitions; ALDH: Aldehyde dehydrogenase.
BCSCs microenvironment
Accumulating evidence has revealed that the TME has a major role in chemo- and radioresistance properties, which is mediated through inflammatory cytokines that help in regulating the self-renewal property of CSCs and maintain cancer progression and metastasis[34,61]. The TME is a mosaic of immune cells such as macrophages,cancer-associated fibroblasts and endothelial cells along with the normal stromal and cancerous cells and the components of the extracellular matrix[54]. These cells present in TME secrete factors such as PDGF, IGF, cytokines such as hepatocyte growth factor and CCL2 leading to the modulation of various signaling pathways, acting in a paracrine or juxtracrine mode that include, TGF-β, Notch, Wnt and Hh that regulate tumorigenesis[55,57].
Macrophages can display either tumor-suppressing or tumor-promoting activity that is dependent on its surrounding microenvironment. Transitioning between the M1 (classical) and M2 phenotype (alternatively activated) can influence the ability of cancer cells to acquire stemness features[62]. Tumor associated macrophages have been correlated with a poor prognosis. M2 macrophages contribute to tissue remodeling/repair, angiogenesis, immunosuppression and tumor progression by secreting pro-angiogenic factors, enzymes and MMPs and promote EMT and hence favor the formation of BCSCs. Notch signaling is required for the activation of macrophages and their differentiation, and hence elevated Notch signaling promotes BCSC formation. M1 macrophages secrete tumoricidal cytokine interleukin (IL)-12,while the M2 tumor associated macrophages secrete the tumor promoting IL-10 cytokine. The hypoxic TME induces the expression of HIF-1, which stimulates the expression of CD47. This cell surface marker, CD47, is enriched in BCSCs and binds to signal regulatory protein α on macrophages to block phagocytosis, whereas CD73 and programmed cell death-ligand 1 (PD-L1) mediate independent mechanisms of evasion of cytotoxic T lymphocytes[63].
The Notch/Jagged1 signaling leads to a positive feedback loop that amplifies cytokine secretion and leads to increased recruitment of M2 macrophages in the tumor[64,65]. Phenotypically, BCSCs with CD44+/CD24-expression in HER2 overexpressing tumor cells display increased EMT potential[66,67], re-emphasizing that EMT serves as a crucial contributor of metastatic potential[68,69]. Studies have shown that treatment of cancer cells with TGF-β, potent inducer of EMT, leads to an increase in CD44+/CD24-/Lowexpressing cells[70], indicative of BCSC phenotype; In addition, STAT3 signaling is upregulated in HER2 cancers and leads to the transition as well as growth of BCSCs[71].
For the survival and maintenance of CSCs, cell-cell and cell-matrix interactions are a necessary requirement (Figure 4), and in this regard cancer cells secrete various factors such as tenascin C, which upregulates Wnt and Notch signaling[72]. CSCs also promote the stromal fibroblasts to secrete periostin, which upregulates Wnt signaling and is necessary for BCSC maintenance[73].
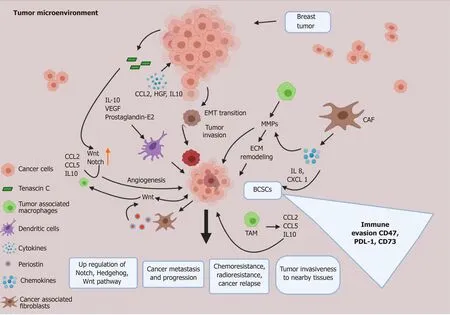
Figure 4 Tumor microenvironment and breast cancer stem cell crosstalk. The tumor microenvironment (TME) plays a critical role in the development as well as the maintenance of breast cancer stem cells. Various signaling molecules secreted by the normal and transformed cells of the TME, such as cancer associated fibroblasts, M2 tumor associated macrophages, endothelial cells and other immune cells, act in a paracrine manner to influence the breast cancer stem cells, which in turn use a positive feedback system to sustain the cells of the TME. In this way the TME and breast cancer stem cells work in a concerted manner to support stemness, chemoresistance, immune evasion and metastatic potential of breast cancer. CAF: Cancer associated fibroblasts; IL: Interleukin; VEGF: Vascular endothelial growth factor; EMT: Epithelial mesenchymal transitions; BCSCs: Breast cancer stem cells; TAM: M2 tumor associated macrophages; PD-L1: Programmed cell death-ligand 1; ECM: Extracellular matrix; HGF: Hepatocyte growth factor.
In BCSCs, chemokines are responsible for the invasive phenotypes as they promote survival, EMT, chemo- and radio-resistance and secretion of MMPs leading to cancer cell invasion[74].
The TME in which the BCSCs reside reveal an intricate interplay of microenvironmental signals that give rise to heterogeneity amongst the CSCs, leading to various subtypes of CSCs with varying EMT phenotypes[75].
Immunofluorescence studies of BCSCs have shown that different subtypes of BCSCs can localize to distinct tumor sites of tissue[76], and CD24-/CD44+cells were found to be localized in invasive sites of tumors with mesenchymal BCSCs features[77],whereas ALDH1+cells displayed proliferative epithelial and mesenchymal features and were present in the interior of tumor stroma[78]. Interestingly, in different types of TNBCs the presence of CSCs having distinctive properties has been seen, which could account for the aggressive nature of these types of tumors. In the claudin-low TNBC tumors, the CSCs are primarily of the mesenchymal phenotype and are CD44+/CD24−/Low, whereas the basal-like TNBCs contain a mosaic of lower numbers of mesenchymal CSCs, but the epithelial ALDH1+BCSCs are more in number[62].
Hence, TME maintains an indispensable cross talk favoring these CSCs.
BCSC RESISTANCE TO FIRST-LINE THERAPY
Various studies conducted over the past decade have shown that acquired resistance to existing conventional chemotherapies is a major challenge faced by clinicians when treating cancer patients. Based upon the biological heterogeneity of BCa, coupled with intratumoral heterogeneity, which can be attributed to the presence of BCSC subtypes,first-line treatment consisting of conventional chemo- and radiotherapy has resulted in the development of CSC clones having the mutations that allow them to survive and continue the tumorigenic process[79].
Chemoresistance in BCSCs
Clinical data collected over the years, has shown that relapse following cytotoxic chemotherapy in BCa patients can be attributed to an increase in the tumor initiating or CSC pool in the tumor and is a cause of treatment failure. Preclinical studies conducted in BCa mammary models have shown an increase in CD44+/CD24-BCSC phenotype population and mammosphere growth following treatment with taxanes like paclitaxel, which are drugs that target microtubules, resulting in a blocking of cell growth followed by apoptosis[80]. Docetaxel treatment also led to a similar pattern resulting in enhanced growth and survival of BCSCs[33]. These results indicate that patients with BRCA1 mutation in hormone-negative cancers are less likely to respond to taxane therapy compared with non-BRCA1 mutation carriers. Therefore, BRCA1 status could be a potential and crucial factor while designing treatment modalities[81].CSCs in an autocrine manner secrete specific cytokines, which in turn promote their own survival and increase the pool of chemoresistant BCa cells[82]. IL-6 is hyperactivated in various cancers and promotes the expression of multiple genes, such as HIF-1 through STAT3 signaling, thereby modulating BCa drug resistance resulting in a poor clinical prognosis[83]. Similarly, overexpression of IL-8 in TNBC leads to transition of cells to the mesenchymal phenotype, stemness and chemoresistance. Compelling evidence has shown the presence of a cytokine loop following drug treatment, which further activates the canonical Wnt and NF-kB pathways.
These results point to the critical role of cytokines in the development of stemness leading to multidrug resistance in BCa, and therefore therapeutics that target these cytokines could provide new strategies for clinicians to improve the treatment outcome in BCa patients.
Radio resistance in BCSCs
Currently, radiotherapy is a standard therapeutic regimen adopted for BCa patients and has shown improved patient survival. However, certain patients do not respond owing to resistance to radiotherapy. The molecular mechanisms resulting in radiation resistance remains poorly understood. Several studies have assessed the effect of radiotherapy on BCSCs and proposed that BCSCs showed survival and enrichment following irradiation[84,85]. Several putative mechanisms have been put forward to account for this radioresistance seen in CSCs, which include enhanced DNA repair capacity, effective scavenging of free radicals formed in response to radiation and the ability to self-renew[85,86]. Another study has given evidence that CSCs normally do not undergo senescence due to the reduced p21 expression coupled with high telomerase activity. BCSCs have been shown to increase the expression of ataxia telangiectasia mutated protein signaling, which has a role in DNA repair checkpoints,hence ataxia telangiectasia mutated activation imparts radioresistance and targeted inhibitors could potentially decrease this resistance to treatment[87].
DRUG TARGETING OF DYSREGULATED BCSC PATHWAYS-CLINICAL INSIGHTS AND FUTURE DIRECTIONS
Advances in early diagnosis and treatment of BCa patients has resulted in a dramatic overall survival rate over the past 10 years. However, BCa is still among the leading cancer-related causes of deaths amongst women worldwide, mainly because of tumor recurrence and resistance to therapy[88]. Distinct BCa molecular subtypes, based on gene expression analysis and hormone receptor presence or absence status, has a defining role in how these subtypes respond to treatment as well as the prognosis.Among the subtypes, TNBC has an extremely poor overall survival and is the most aggressive subtype; this is linked to the presence of a higher percentage of CSCs having CD44+/CD24-phenotype with stem cell-like and invasive features compared to other cancer subtypes[89,90]. Whereas, luminal and HER2+subtypes are known to be CD44+/CD24-and ALDH1+[91,92].
As reported earlier the ALDH1 enzyme, which is highly expressed in BCSCs,catalyzes the oxidation of various substrates and toxins. The molecular disparities in terms of CD44, CD24 and ALDH1 CSC markers expression in distinct BCa histological subtype cohorts is the plausible reason for the observed BCa outcomes. In addition,BCSCs have been reported to have an altered redox potential that helps in the detoxification processes as well as regulates the levels of reactive oxygen species and hence promotes their viability. Strategies that can target this redox state in BCSCs have shown a phenotypic switch from the chemoresistant mesenchymal cell type to the chemosusceptible epithelial one[93].
This knowledge is important to narrow down the treatment regimens for different subtypes. Hormone dependent types respond to ER blockers (tamoxifen, fulvestrant),and HER2 receptor blockers (Herceptin). However, in the clinically difficult to treat TNBC patients, hormonal or anti-HER2 therapies do not show any response and hence poly (ADP-ribose) polymerase inhibitors and monoclonal antibodies directed against PD-L1 have been approved[94].
The major challenge currently is to prevent metastasis, therapy resistance and relapse of the disease. As per current data, BCSCs are the key drivers responsible for regenerating tumors and helping in survival of existing tumors. However, targeting of these cells is difficult as they form a part of the tumor niche, supported by various factors promoting their survival in the TME. Hence, interaction of BCSCs with cellular and non-cellular components present as a part of TME can be selected as an important target for therapeutic approaches[95].
The higher resistance to conventional therapy observed in BCSCs compared to other cells in the tumor bulk necessitates the development of more specific treatment modalities. Aberrations in Notch, Hh and Wnt pathways have been implicated to cause tumorigenesis in mammary glands, and therapies against the above-mentioned pathways could show promise in treating these BCSC population[96,97].
Notch pathway
The clinically acceptable approach to inhibit this pathway is through the action of γsecretase inhibitors (GSIs), which prevent the cleavage of the Notch receptor thereby blocking the release of Notch intracellular domains. Normally the Notch intracellular domains translocates to the nucleus where it interacts with other cofactors, initiating gene transcription. MK-0752, a GSI developed by Merck has reached phase I/II trials in combination with docetaxel for metastatic BCa. Biopsies from patients enrolled in the trial showed a decreased CD44+/CD24-population, ALDH+activity and reduced stem cell population, hence validating that specific targeting of a BCSC pathway is effective along with systemic therapy in treating cancer[98]. RO4929097, another GSI,decreased Notch intracellular domains levels and led to drastic reduction in the expression of the Notch target genes.
Wnt pathway
The monoclonal antibodies, vantictumab (OMP-18R5) and cirmtuzumab (UC-961),anti-Frizzled and anti-ROR1 were assessed in combination with paclitaxel for metastatic BCa. A clinical trial is currently underway by Novartis Pharmaceuticals in the United States and Spain to find the recommended dose of LGK974 as a single agent and in combination with the humanized antibody PDR001 that can be safely given to adult patients suffering from various cancers including aggressive, hard-totreat BCa[99].
Hh pathway
Activation of Hh signaling is linked to overexpression of MDR1 and ABCG2 in BCSCs,pointing to the need of inhibiting this pathway to prevent resistance to first-line treatment. Clinical trials using small molecule antagonists of the Hh pathway that bind to Smo, such as Vismodegib can inhibit the amplified signaling seen in aggressive,metastatic TNBCs along with conventional interventions (NCT02694224). Sonidegib(LDE225) a potent and selective Smo inhibitor along with cisplatin and etoposide were effective in patients with Sox2 activation and have been tested in phase I/II clinical trials BCa trial (NCT02027376).
Other trials using Smo inhibitors in TNBCs included LDE225 (NCT01757327) and combined treatment with RO4929097 (GSI of the Notch pathway) and vismodegib(NCT01071564). Sims-Mourtada and coworkers tested Hh inhibitors along with docetaxel that revealed a superior effect by decreasing the CD44+/CD24-BCSC population and mammosphere formation as compared to treatment with docetaxel alone[100].
Drugs targeting other pathways
Various proteins can give a survival and growth advantage to the BCSCs and in this regards the role played by myeloid cell leukemia 1, anti-apoptotic protein Survivin,cyclin D1 and D3 (cell cycle related), Oct4, Sox2 and Nanog have been explored.Pharmacological strategies to target the enzyme eukaryotic initiation factor 4A(eIF4A1), which plays a critical role in unwinding mRNA secondary structures in the 5’-untranslated region and allows the binding of the small ribosomal subunit to initiate protein translation, can effectively downregulate/silence the expression of all the above mentioned genes[101] and prevent the relapse. A clinical trial (NCT04092673)for chemoresistant tumors including BCa is underway to look into the antitumor activity of Zotatifin (eFT226), a small molecule that by inhibiting eIF4A1 downregulates RTK expression and inhibits PI3K/AKT and MAPK signaling.
In normal and CSCs the Hippo pathway acting through the transcriptional coactivators Yap and Taz promotes tissue proliferation and self-renewal leading to metastasis. Elevated mevalonate pathway has been shown to upregulate of the Hippo pathway, and hence clinical trials using inhibitors of the mevalonate pathway such as Zoledronate, a bisphosphonate, which could interfere with YAP/TAZ expression,(NCT03358017, NCT02347163) have been carried out. Another clinical trial(NCT02370238) to look into the efficacy of Reparixin, an inhibitor of CXC chemokine receptor types 1 and 2 to specifically target BCSCs in HER2 amplified TNBC has shown promise. Bevacizumab, which specifically targets vascular endothelial growth factor signaling (NCT01190345), has been shown that in combination with chemotherapy, an improved response rate, and progression-free survival was seen in advanced metastatic BCa, possibly by targeting BCSCs[102].
Bivatuzumab (humanized monoclonal antibody) mertansine (NCT02254005) has completed a phase I trial for metastatic BCa targeting stem cell surface marker CD44 v6.
CDK inhibitors, which could lead to a senescence phenotype and favorable outcomes in advanced, metastatic BCa by targeting the BCSCs, are being explored.Studies reveal that targeting cyclin D1 or CDK4/6, inhibition in the invasive ability of BCSCs was noted, due to the dysregulated cell cycle operative in these cells. Data mining using high-end bioinformatic tools from The Cancer Gene Atlas revealed that 32 genes correlated with the development of stem cell signatures in the various subtypes of BCa, such as luminal A and B and TNBC[103].
A large number of clinical trials for the efficacy of Palbociclib, a CDK 4/6 inhibitor(NCT02738866, NCT03709082, NCT02491983) in conjunction with endocrine and aromatase therapy for metastatic BCa are underway and show promise.
The ability of the ionophore salinomycin to target the upregulated nucleolin protein in BCa has revealed targeted suppression of BCSCs[104]. Furthermore, 4LB5, a recombinant drug specifically targeting nucleolin, has shown promise in the management of TNBC[105] (Table 2 and Figure 5).
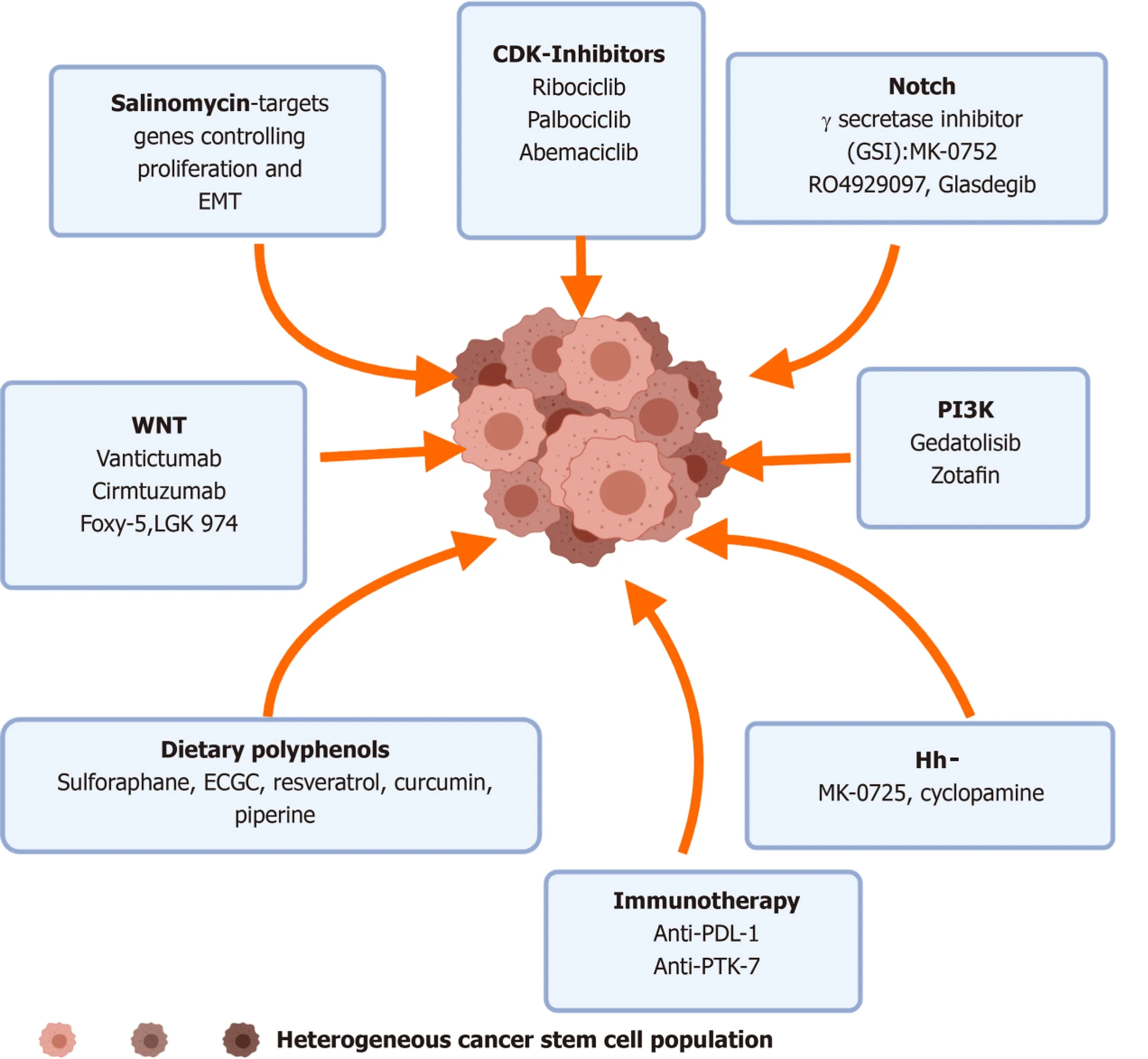
Figure 5 Pharmacological targeting of breast cancer stem cells. Insights from preclinical and clinical studies have shown that various small molecular inhibitors and antibodies against the effectors of the aberrantly expressed pathways can be targeted to downregulate the stemness and altered phenotype of breast cancer stem cells. EMT: Epithelial mesenchymal transitions; CDK: Cyclin dependent kinase; GSI: γ-secretase inhibitors; PI3K: Phosphatidylinositol 3-kinase; PTK:Protein tyrosine kinase.
Immunotherapy against BCSCs
CSCs have the ability to evade and to suppress the immune system, and reports have shown that BCSCs have low expression of the MHC class I cell surface receptor and secrete various cytokines (IL-10, IL-4, TGF-β) that block the activation of immune T and natural killer cells[106]. Furthermore, the elevated expression of PD-L1 on BCSCs is another means by which these cells avoid eradication by the immune cells[107]. Anti PD-L1 therapy as a means to lower the levels of this immune checkpoint, which is overexpressed in BCSCs, are being conducted. Antibodies against PD-L1 (MEDI4736)used in combination with other chemotherapeutic drugs are being used in over 23 registered trials of TNBC (NCT02685059, NCT02489448).
The conundrum lies in the ability to specifically target markers present on BCSCs rather that the MaSCs or differentiated noncancerous cells. Experimental preclinical and clinical studies are underway to look for novel targets in the CSC population. A feasibility study in BCa patients (NCT02831634) for a method to identify tumor mutated epitopes MAGE-A expressed on BCSCs and T-cell responses to neo-epitopes is underway. A preclinical study carried out in breast tumor bearing mice for efficacy of vaccines against cell surface Cripto-1 protein, which is highly expressed in BCSCs from Her2+ and TNBC subtypes and leads to their self-renewal, has shown exciting results for migration and EMT[108].
CONCLUSION
The current status of research on BCSCs reveals that tumorigenesis and BCa progression are not stand-alone processes, rather are highly coordinated and interlinked and are regulated by many active molecules and genes acting through several interconnected pathways. BCSCs as a new and promising paradigm for BCa treatment requires focused and robust data that specifies the key genes involved for progression of these CSCs population, which is a major reason behind disease relapse and treatment resistance (Figure 6).
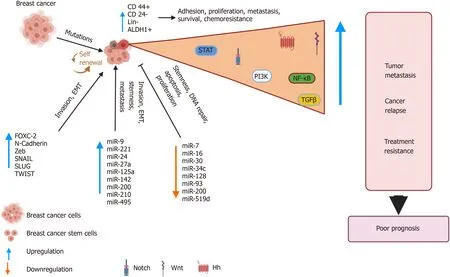
Figure 6 Breast cancer stem cells: drivers of tumorigenesis and cancer relapse. Genomic instability due to inherent mutations or in response to treatment results in alterations in various pathways. This abnormal signaling drives the development of a distinct subpopulation of breast cancer stem cells (BCSCs),which display characteristic cell surface markers and express genes that allows unlimited self-renewal and differentiation abilities. The BCSCs also display altered microRNA profiles that are used by the BCSCs to invade, metastasize and acquire stemness features. The net result is the development of an aggressive, treatment resistant tumor, which results in relapse. BCSCs: Breast cancer stem cells; NF-κB: Nuclear factor kappa beta; PI3K: Phosphatidylinositol 3-kinase; TGFβ:Transforming growth factor beta; EMT: Epithelial mesenchymal transitions; Hh: Hedgehog; ALDH: Aldehyde dehydrogenase.
The TME, a complex network of cellular and acellular factors, secrete various cytokines that promote the survival and expansion of the CSC pool. By devising strategies that can interfere with the BCSC-cytokine axes, improvement in clinical outcomes could evolve in the near future. In this regard, modulation of TME for BCSC targeting would need to be effectively shown in clinical trials with a provision of following up the patients for a longer duration to show long-term tumor-free survival.
Further refinement in systemic therapy and more stringent clinical trials to develop specific treatment against BCSCs and target their growth and tumorigenicity are also warranted. To further complicate issues, heterogeneity also exists within these BCSCs,which limits therapeutic efficacy. Strategies that could effectively deliver tumor suppressing miRNAs or silence the oncogenic miRNAs in various BCSCs subtypes are the need of the hour. The contribution of drugs to the development of the stemness phenotypes in the breast tumor stroma requires novel and innovative methods to avoid induction of mutational changes, reduce toxic effect causing development of stem cells and also limit the toxicity to normal cells.
Advances in therapeutics that could take advantage of combination therapies and molecular profiling for the eradication of BCSCs could potentially become a promising strategy for patients diagnosed with BCa.
ACKNOWLEDGEMENTS
We are grateful to institutional support provided by Amity University, Noida, India.We are thankful to Biorender.com for the preparation of figures.
杂志排行

World Journal of Stem Cells的其它文章
- Epigenetic modulators for brain cancer stem cells: Implications for anticancer treatment
- Mechanisms involved in selecting and maintaining neuroblastoma cancer stem cell populations, and perspectives for therapeutic targeting
- Roles of mitochondrial unfolded protein response in mammalian stem cells
- Stem cell therapies in tendon-bone healing
- Exosomal microRNAs from mesenchymal stem/stromal cells:Biology and applications in neuroprotection
- Immunotherapy against programmed death-1/programmed death ligand 1 in hepatocellular carcinoma: Importance of molecular variations, cellular heterogeneity, and cancer stem cells