Immunotherapy against programmed death-1/programmed death ligand 1 in hepatocellular carcinoma: Importance of molecular variations, cellular heterogeneity, and cancer stem cells
2021-07-30CaeciliaSukowatiKorriElvanitaElKhobarClaudioTiribelli
Caecilia H C Sukowati, Korri Elvanita El-Khobar, Claudio Tiribelli
Caecilia H C Sukowati, Claudio Tiribelli, Centro Studi Fegato, Fondazione Italiana Fegato ONLUS, Trieste 34149, Italy
Korri Elvanita El-Khobar, Hepatitis Unit, Eijkman Institute for Molecular Biology, Jakarta 10430, Indonesia
Abstract Hepatocellular carcinoma (HCC) is a heterogeneous malignancy related to diverse etiological factors. Different oncogenic mechanisms and genetic variations lead to multiple HCC molecular classifications. Recently, an immune-based strategy using immune checkpoint inhibitors (ICIs) was presented in HCC therapy,especially with ICIs against the programmed death-1 (PD-1) and its ligand PD-L1.However, despite the success of anti-PD-1/PD-L1 in other cancers, a substantial proportion of HCC patients fail to respond. In this review, we gather current information on biomarkers of anti-PD-1/PD-L1 treatment and the contribution of HCC heterogeneity and hepatic cancer stem cells (CSCs). Genetic variations of PD-1 and PD-L1 are associated with chronic liver disease and progression to cancer. PD-L1 expression in tumoral tissues is differentially expressed in CSCs,particularly in those with a close association with the tumor microenvironment.This information will be beneficial for the selection of patients and the management of the ICIs against PD-1/PD-L1.
Key Words: Hepatocellular carcinoma; Programmed death-1; Programmed death ligand 1;Cancer stem cells; Cancer heterogeneity; Genetic variants
INTRODUCTION
International epidemiology data Globocan 2018 predicted primary liver cancer to be the sixth most commonly diagnosed cancer and the fourth leading cause of cancerrelated mortality worldwide. In the male population, its incidence and mortality were 2 to 3 times higher compared to females, ranking it as fifth in terms of global cases and second in terms of deaths[1]. Hepatocellular carcinoma (HCC) accounts for about 90%of liver cancer cases, with cirrhosis as the strongest underlying condition[2,3].
HCC is caused by various etiological factors. Major risk factors for HCC are liver cirrhosis due to chronic hepatitis B virus (HBV) and/or hepatitis C virus (HCV)infection, which comprised around 80% of HCC cases globally[4], alcoholic liver disease (ALD), non-alcoholic fatty liver disease (NAFLD) leading to non-alcoholic steatohepatitis (NASH), and exposure to aflatoxin B1. It is noteworthy that even though chronic HBV and HCV infection is the current major driver of HCC cases, the rise of liver disease due to metabolic syndrome (NAFLD/NASH) may lead to a high number of HCC cases in the future[5,6].
The international consensus for HCC management[7] recommends surgical intervention as the main curative treatment for HCC, resulting in the best outcomes in well-selected candidates (five-year survival of 60%-80%)[8]. Image-guided radiofrequency ablation is the treatment of choice for HCC patients with early-stage HCC when liver transplantation or hepatectomy are precluded. For patients in an intermediate stage, palliative treatment using trans-arterial chemoembolization(TACE) is recommended[7,9]. For advanced HCC, oral systemic treatment with the tyrosine-kinase inhibitor sorafenib may extend the patient’s overall survival (OS) for around 3 mo[10]. Despite the fast emergence of targeted therapy development, HCC remains largely incurable due to low response rate and therapeutic resistance[11].
PROGRAMMED DEATH-1/PROGRAMMED DEATH LIGAND 1 IMMUNE CHECKPOINT INHIBITORS
Immunotherapy represents an effective and promising option against various types of cancer. Recently, a new immune-based strategy using immune checkpoint inhibitors(ICIs) for HCC therapy was shown to be highly promising compared to chemotherapy and systemic therapy. Immune checkpoints are pathways that inhibit the immune response to maintain self-tolerance and regulate the duration and amplitude of immune responses[12]. The liver tissue is immune tolerant due to its physiological function, and liver sinusoidal endothelial cells are exposed to a significant amount of bacterial antigens from the portal circulation[13].
ICIs against the programmed death-1 (PD-1, CD279) and its ligand, is an important focus in cancer immunology and oncology with FDA approval for various types of cancer. Immunotherapies targeting PD-1/programmed death-ligand 1 (PD-L1)signaling have now become the first-line treatment for some cancers due to their promotion of anti-tumor immune responses in patients with advanced cancers[14].
PD-1 is a cell surface receptor belonging to the extended CD28/CTLA-4 (cytotoxic T-lymphocyte-associated protein 4) family. It is an approximately 55-kDa type I transmembrane glycoprotein that exists as a monomeric receptor. PD-1 is mainly expressed on T cells, B cells, monocytes, dendritic cells (DCs), and natural killer (NK)cells[15,16]. In a phase I/II CheckMate 040 trial in 2017, nivolumab, a checkpoint inhibitor against anti-PD-1 showed promising results in advanced HCC patients[17]. It was well tolerated and patients had a long-lasting response. Nivolumab is a human immunoglobulin G4 (IgG4) monoclonal antibody that binds to the PD-1 receptor and disrupts the interaction with PD-L1 and PD-L2, its ligands in tumor cells. This interaction releases PD-1 pathway-mediated inhibition of the immune response,including the anti-tumor immune response[18]. Other ICIs, pembrolizumab (anti PD-1)[19] and tremelimumab (anti CTLA)[20,21] were under phase 2 trial both as single or as combination therapy. It was shown that anti-PD-1 therapy in HCC patients intolerant to sorafenib resulted in an excellent complete response[22].
A more recent approach is to target the ligand PD-L1. PD-1 has two ligands from the B7 transmembrane proteins family, the PD-L1 (B7-H1) and PD-L2 (CD273, B7-DC)[16]. PD-L2 affinity to PD-1 is three-fold higher than PD-L1; however, PD-L2 is only expressed in antigen-presenting cells[15]. PD-L1 is a 40-kDa type I transmembrane protein, which is expressed in immune cells (ICs) such as T cells, B cells, NK cells, DCs,macrophages, and myeloid-derived suppressor cells. It is also expressed in non-IC types including epithelial, endothelial, and tumor cells[14,23].
The safety and activity of PD-L1 inhibition using the engineered humanized antibody atezolizumab was first reported in lung cancer[24,25]. It is a high-affinity human monoclonal IgG1 antibody that specifically binds to PD-L1 and prevents its interaction with PD-1 and B7.1[26]. In HCC, a phase 1b GO30140 study of atezolizumab plus bevacizumab (a monoclonal antibody against VEGF) in untreated patients with unresectable HCC showed an acceptable low side-effect profile and promising antitumor activity with a median progression-free survival (PFS) of 7 mo[27]. Recently, the IMbrave150 study, a global, multicenter, open-label, phase 3 randomized trial, demonstrated the safety and efficacy of atezolizumab plus bevacizumab as compared with sorafenib. In 501 unresectable HCC patients at 111 sites in 17 countries, PFS was significantly longer with atezolizumab–bevacizumab than with sorafenib with a median of 6.8vs4.3 mo. The OS was also significantly longer in this group with the estimated rates of survival at 12 mo was 67%vs55%[28].
However, despite the success of ICIs against the PD-1/PD-L1, a substantial proportion of patients fail to respond. In 29 unresectable HCC patients, the objective response rate after nivolumab monotherapy was around 21% with an OS of 26 wk.Interestingly, differential responses to nivolumab among multiple tumor nodules in a single HCC patient were found in 18% of total cases, where small metastatic tumors but not large tumors regressed[29]. The efficacy of immunotherapy might depend on different factors, such as patients’ heterogeneity on genomic features, oncogenic pathways, cancer microenvironment, systemic immunity status, microbiome, and metastases, as reviewed in[30].
PD-1/PD-L1 SIGNALING PATHWAYS
PD-1 transcription may be induced by various transcription factors including the nuclear factor of activated T cells, NOTCH, Forkhead box protein (FOX) O1, and interferon regulatory factor 9 (IRF9)[31]. During antigen stimulation, binding of PD-1 and its ligand leads to phosphorylation of the two PD-1 tyrosine residues, the immunoreceptor tyrosine-based inhibitory motif (ITIM) and immunoreceptor tyrosine based switch motif (ITSM), in the PD-1 cytoplasmic region, followed by recruitment of SHP1 and SHP2 phosphatases to ITIM and ITSM, respectively[32,33]. These interactions resulted in decreased phosphorylation of various signaling molecules including ZAP70/CD3 and downstream PKC signaling, vav, AKT, and ERK in T cells,and Ig and ERK in B cells[34,35].
PD-L1 is activated by pro-inflammatory cytokines such as IFN-γ and IL-4, through its IFN regulatory factor 1 (IRF1) response element in the PD-L1 promoter region[36,37]. PD-1 interaction with either ligand has been shown to inhibit T cell proliferation through cell cycle arrest at G0/G1, promote apoptosis, and stimulate immunosuppressive IL-10 secretion but impair IL-2 secretion[35,38,39]. PD-L1 also regulates macrophage proliferation and activation. PD-L1 negatively regulated macrophage proliferation, survival, and activation by inhibition of the mTOR pathway, resulting in an immunosuppressive phenotype. However, treatment with anti-PD-L1 antibody, but not anti-PD-1 antibody, reversed this phenotype and triggered macrophage-mediated anti-tumor activity instead[40].
The PD-1/PD-L1 axis plays an important role in successful protective cellular immune response and prevention of immune overstimulation and autoimmune disorders through maintenance of T cell homeostasis and control of self-tolerance[14,36,38] (adapted in Figure 1). PD-L1 binding to PD-1 leads to the inhibition of T cell functional activation by: (1) Directly targeting the T cell receptor (TCR) signaling through phosphatases recruitment and inhibition of ZAP70 and PI3K downstream pathways; (2) Indirectly inhibiting TCR signaling and T cell proliferation by regulating CK2 expression and activation; (3) Regulating TCR surface expression by promoting E3 ubiquitin ligases expression, leading to TCRs removal from the T cell surface and loss of T cell response upon antigen stimulation; and (4) Altering T cell metabolism by inhibition of ERK and PI3K/Akt activities, resulting in a long-lived memory T cells phenotype[32,34]. This PD-1 TCR-dependent inhibition of T cell activation occurs with or without CD28 or ICOS (inducible T cell co-stimulator) co-stimulation, although CD28 co-stimulation reduced PD-1 efficiency in inhibiting TCR-dependent T cell activation[34].
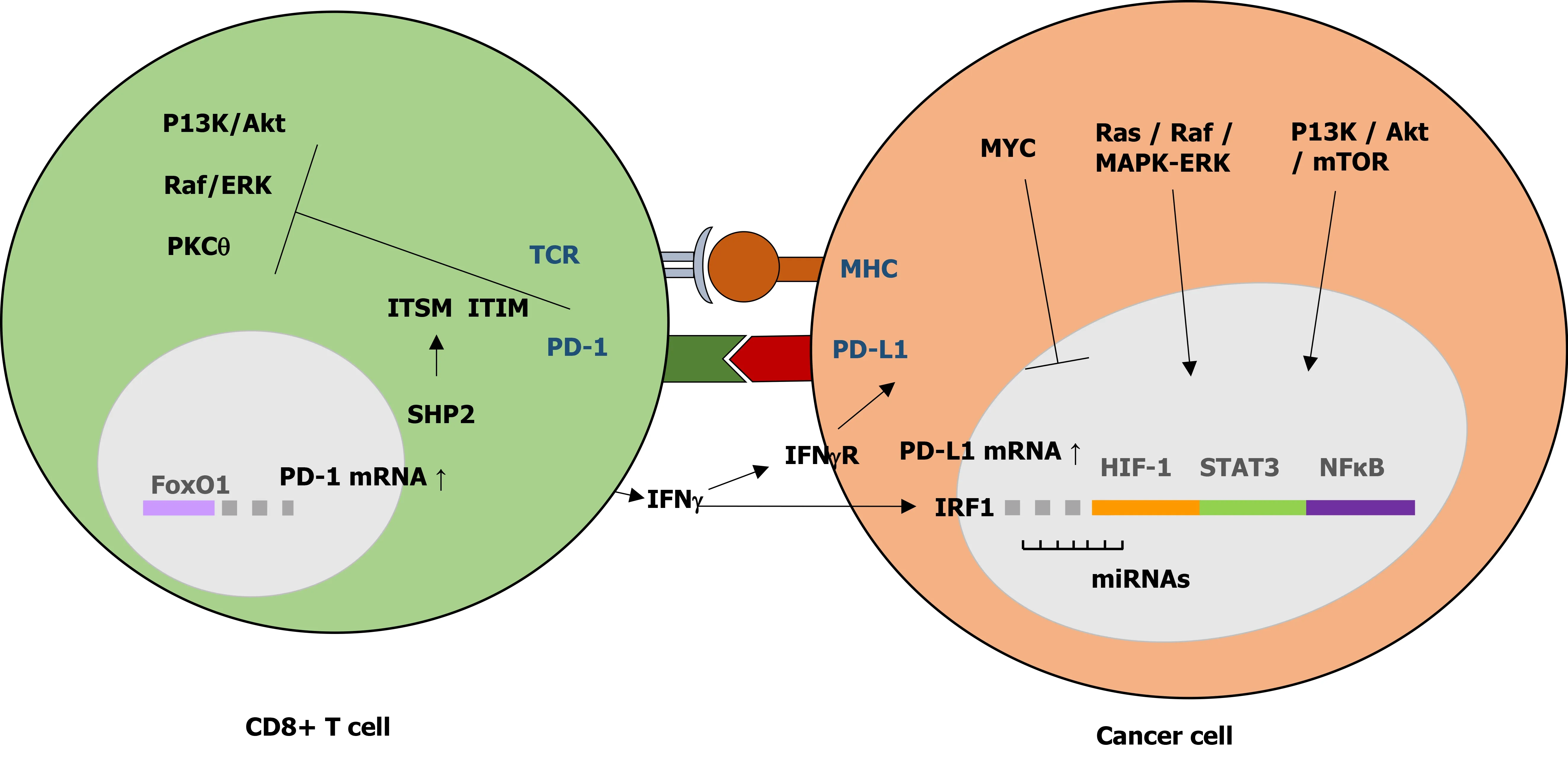
Figure 1 Regulatory mechanism of programmed death-1/programmed death ligand 1 signaling in cancer. The interaction between programmed death-1 and programmed death ligand 1 (PD-L1) leads to the inhibition of T cell functional activation. PD-L1 expression in cancer cells is regulated by aberrant oncogenic pathways. PD-1: Programmed death-1; PD-L1: Programmed death ligand 1; TCR: T cell receptor; ITSM: Immunoreceptor tyrosine-based switch motif;ITIM: Immunoreceptor tyrosine-based inhibitory motif; IRF: Interferon regulatory factor.
The PD-1/PD-L1 dependent T cell activation is often found to be dysregulated or overexpressed in tumor cells and viral-infected cells. Overexpression of PD-1/PD-L1 in tumor and viral-infected cells promotes self-tolerance by inducing PD-1 dependent inhibitory signals, just like in normal cells, thereby escaping T cell-mediated immune response and promoting tumor progression and survival[15].
High PD-L1 expression in cancers is associated with a poor prognosis. Cancer cell’s PD-L1 binding to PD-1 receptor on tumor-infiltrating T cells (TILs) has been shown to induce SHP2 activation resulting in suppression of the TCR pathway and inhibition of T cell activity[41]. During viral infection, PD-1 is expressed transiently by CD8+T cells,which will gradually decline at the end of acute infection due to lack of specific TCR stimulation[15]. However, in chronic viral infection, exhausted CD8+T cells maintained PD-1 expression due to continuous TCR-antigen ligation and lack of PD-1 promoter re-methylation[15,42]. The lack of DNA re-methylation left the PD-1 locus ready for rapid PD-1 expression, providing a premature termination of CD8+T cell antiviral functions[42].
PD-1/PD-L1 overexpression in many cancers causes functionally exhausted T cells,thus blocking the PD-1/PD-L1 pathway may restore anti-tumor immunity by enhancing T cell killing activities and improve cancer prognosis[41]. In addition, PD-1 has been shown to bind and phosphorylate two mTOR downstream effectors,eukaryotic initiation factor 4E (eIF4E) and ribosomal protein S6 (rpS6), to promote tumor growth in HCCs[43].
PD-L1 expression in cancer is complex. It is regulated differently at transcriptional and post-transcriptional levels. PD-L1 can be activated by aberrant oncogenic signaling pathways including Ras/Raf/MEK/MAPK-ERK and PI3K/Akt/mTOR, and various transcription factors such as STAT-3, STAT-1, c-Jun, HIFs, and NF-κβ . PD-L1 was also regulated post-transcriptionally by various microRNAs (miRNAs), which bind to PD-L1 mRNA to either repress or enhance translation[16]. The binding of miR-4717 and miR-570 to PD-L1 3’UTR resulted in downregulation of PD-L1 mRNAs[44,45]. Several recent studies have also reported additional regulatory mechanisms of PDL1 expression in cancer, including epigenetic regulation through methylation and histone acetylation, post-translational modification on PD-L1 protein including phosphorylation, N-glycosylation, poly-ubiquitination, and palmitoylation, and various genetic alterations[41]. PD-L1 expression in HCCs was epigenetically regulated by EZH2-induced upregulation of H3K27me3 levels on the PD-L1 promoter and IRF1 promoter, an essential transcription factor PD-L1, without affecting the IFNγ-STAT1 pathway[46]. In addition, PKM2-induced phosphorylation of histone H3 was important for EGF-mediated PD-L1 transcription in HCC[47].
Oncogenic Ras signaling upregulated PD-L1 expression in tumor cells through modulation of the AU-rich element-binding protein tristetraprolin (TTP), located in the 3’UTR of PD-L1 mRNA, resulting in stable PD-L1 mRNA expression. Furthermore,MEK signaling, Ras downstream effectors, phosphorylated and inhibited TTP activity through kinase MK2 activation[48]. Downregulation of PD-L1 expression in liver cancer cells was the result of Tyr56 phosphorylation and activation of glycogen synthase kinase 3β (GS3Kβ), part of the Wnt/ β-catenin pathway, by MET (hepatocyte growth factor) receptor tyrosine kinase. Treatment with MET inhibitors in these cells decreased the antitumor activity of T cells in HCCs[49], possibly through PD-L1 effects on CD8+T cell activity.
The oncogene MYC has been shown to affect PD-L1 expression in cancer cells. In HCC, MYC activation was shown to downregulate PD-L1 expression through the reduced level of STAT-1, a crucial part of the IFN-γ pathway[50]. This observation was in line with a previous study which showed a positive correlation of JAK-2 expression,a STAT-1 upstream effector, with increased PD-L1 expression in nodular sclerosing Hodgkin lymphoma and large B-cell lymphoma[51]. p53 tumor suppressor gene is found frequently mutated in many types of cancer. Increased p53 expression was related to increased PD-L1 in tumor cells of HCC and oral squamous cell carcinoma[52,53]. p53 expression in HCCs was also positively correlated with an increased level of APE-1[52], a multifunctional enzyme involved in the base excision repair pathway.Additionally, p53 level was associated with IFN-γ-induced PD-L1 expression in melanoma through maintenance of JAK-2 expression in tumor cells[54].
IFN-γ induces PD-L1 mRNA and protein expression through upregulation of transcription factor IRF-1, and its binding to the interferon stimulated response element in the PD-L1 gene promoter. This IRF-1 upregulation of PD-L1 expression is antagonized by IRF-2 competitive binding to the PD-L1 promoter[55]. Increased IRF-1 mRNA expression can be observed in patients with well-differentiated or early stages of HCC tumors[55]. IFN-γ upregulation of PD-L1 was associated with epithelial to mesenchymal transition (EMT) in pancreatic ductal adenocarcinoma (PDA), characterized by increased vimentin expression and infiltration of CD8+T cells and Foxp3+cells. This EMT promotion of IFN-γ in PDA was inhibited by the treatment of STAT-1 siRNA[56]. In HCC, PD-L1 upregulation also promoted EMT, characterized by increased N-cadherin but reduced E-cadherin levels, through the activation of SREBP-1 of the PI3K/Akt pathway[57,58].
ICs that infiltrate some types of HCC also secrete IFN-γ. CD8+CTLs secretion of IFN-γ is impaired following CD8+CTLs upregulation of PD-L1 in HCC tumor cell lines by HLA class-I specificity, which indicates a negative feedback regulatory mechanism of IFN-γ and PD-L1 expression in CD8+CTLs[59]. Some HCC cells expressed myocyte enhancer factor 2D (MEF2D), which was associated with high PDL1 expression and shorter survival time. MEF2D expression in HCCs also negatively correlated with lower numbers of CD4+and CD8+T cells, attenuating its antitumor activity. IFN-γ treatment of HCC cells resulted in MEF2D acetylation by p300 activation, followed by MEF2D binding to the PD-L1 gene promoter and upregulation of PD-L1. Without IFN-γ, MEF2D acetylation was inhibited by SIRT-7 by forming a complex with MEF2D[60]. Independently, SIRT-7 seemed to promote HCC cell proliferation, as SIRT-7 knockout mice had reduced cell proliferation and tumor growth[60].
Hypoxia commonly occurs in the tumor microenvironment. ICs secrete tumorpromoting inflammatory cytokines under hypoxic conditions, which further activate STAT-3 and NF-κβ transcription factors resulting in tumor proliferation, survival,and invasion[61]. NF-κβ and HIF-1a (hypoxia-inducible factor 1a) were shown to upregulate PD-L1 expression in non-small cell lung carcinoma (NSCLC), through epidermal growth factor receptor activation and phosphorylation of Akt and ERK[62].Increased HIF-1a level in HCCs was also associated with high PD-L1 expression and increased risk of cancer recurrence and metastasis[63]. HIF-1a level in HCC was also correlated with increased CXCL-12 mRNAs, a ligand for CXCR-4 chemokine receptor,which was known for its positive effect on migration, proliferation, and survival of cancer cells[64]. Accordingly, loss of nuclear CXCL-12 expression was correlated with better OS[64]. HIF-1a also induced TREM-1 (triggering receptor expressed on myeloid cells-1) in tumor-associated macrophages (TAMs), which were found abundantly in advanced stages of HCCs, resulting in impaired cytotoxic functions and induced apoptosis of CD8+T cells[65]. Blocking of TREM-1+TAMs led to reversal of its immunosuppressive effect and PD-L1-induced resistance in liver cancer cells[65].
PD-1/PD-L1 EXPRESSION IN HCC
PD-1 expression
Aberrant PD-1/PD-L1 binding which leads to activation of the self-tolerance pathway has been observed in both ICs and tumor cells. Increased PD-1 and PD-L1 expression was observed in pathological liver specimens[66]. PD-1 was mostly noted in tumorinfiltrating CD8+T cells. HCCs with a PD-1-high cell population were aggressive and had higher levels of predictive biomarkers of response to anti-PD1 therapy[67,68]. The PD-1 expression was also elevated in monocytes of HCC patients[69].
High CD8+T cells seemed to result in a favorable outcome for HCC[70]. Low CD8+T cell infiltration in HCCs has been associated with EMT through increased vimentin expression resulting in poor patient prognosis[56]. CD8+T cells in HCCs seemed to express PD-1 at different levels. PD-1-high CD8+T cells which expressed LAG-3(lymphocyte-activation gene 3), a T cell expression marker, and/or TIM-3(T-cell immunoglobulin mucin-3), produced low levels of IFN-γ and TNF in response to anti-CD3. Treatment of this subset of cells with antibodies against PD-1 and LAG-3 or TIM-3 restored both cell proliferation and cytokine production[67] making them more susceptible to immune checkpoint blockade therapy.
A higher ratio of LAG-3+to CD8+cells was found on HCC tissues compared to adjacent normal tissues in a cohort of 143 Chinese patients. A high level of LAG-3 (T cell expression marker expressed on both CD4+and CD8+cells) was also associated with a high level of FGL-1 (fibrinogen-like protein 1), a major ligand for LAG-3, but not PD-L1 level[70]. A previous study in Caucasian HCC patients reported that tumor cells have increased PD-L1+and LAG-3+cells but reduced CD8+T cells expression[71].Taken together, it seems that PD-1, PD-L1, and LAG-3 expression was regulated differently in different HCC subsets, in association with the regulation of CD8+T cell tolerance. However, it can be inferred from all studies that high LAG-3 expression in HCCs, which was associated with poor disease outcome[67,70,71], may represent another layer of tumor evasion mechanism based on its effect on T cell activation.
Tumor-infiltrating ICs (TIICs) play crucial roles in the reactivation of effective antitumor responses[72]. TIICs in HCCs have also been shown to express high LAG-3 positivity[71]. Using advanced cytometry by time-of-flight (CyTOF) analysis, detailed TILs profiling in a spontaneous HCC model that was resistant to anti-PD-1 treatment revealed that effector memory CD8+T cells (CD44+CD62L-KLRGint) had a high level of T cell exhaustion markers, TIGIT (T-cell immunoglobulin and ITIM domain), LAG-3,and CD39. Furthermore, this enhanced TIGIT expression on CD8+and CD4+T cells was tumor specific[73]. In addition, they also found a high level of PVRL1 (poliovirus receptor-related 1) mRNA and protein in HCC tissues. This PVRL1 upregulation stabilized cell surface PVR (poliovirus receptor) which interacted with TIGIT, an inhibitory molecule on CD8+effector memory T cells[73]. Inhibitors of the PVRL1/PVR/TIGIT signaling axis may be beneficial for the development of HCC treatment, in combination with anti-PD-1, due to its induction of the anti-tumor immune response[72,73].
PD-L1 expression in tumor cells
Many tumors overexpress PD-L1 to escape immune surveillance by deregulating the survival and proliferation pathways[74]. Elevated PD-L1 expression has been reported in various cancers and was strongly correlated with advanced disease state and unfavorable prognosis[14]. Blockade of the PD-1/PD-L1 signaling axis by anti-PD-1 and/or anti-PD-L1 antibodies resulted in reactivation of the exhausted ICs in the tumor microenvironment and elimination of cancer cells[41]. In HCC, PD-L1 was stained positive in tumor cells and TILs, but rarely in normal hepatocytes[26]. PD-L1 expression in tumor cells may be constitutive by the regulation of oncogenic events in tumor cells, and inductive, by the stimulation of immune response in the tumor microenvironment[75]. PD-L1 staining showed that the main PD-L1 expression on HCC cells was in the cell membrane with variable staining in the cytoplasm[76-79].
PD-L1 expression was found to be higher in HCC tissue compared to the corresponding non-tumor liver[64,80]. High PD-L1 expression was associated with tumor size and histological grade. Furthermore, Kaplan Meier analysis showed that hepatic membrane-bound PD-L1 expression represented a predictive biomarker for HCC aggressiveness and patient survival[64,76,78], as summarized in a recent meta-analysis of 23 studies with around 3,500 patients[81].
Higher expression of PD-1/PD-L1 was associated with shorter OS and tumor-free survival. Furthermore, circulating PD-1/PD-L1 expression was also closely correlated with intratumoral PD-L1 expression[82]. Overexpression of both PD-L1 and PD-L2 was also observed in resected HCC tissues, which were also associated with poor survival[78]. In addition, increased PD-1 expression in HCC is associated with the promotion of tumor growth[43]. There are several anti-PD-1 and anti-PD-L1 drugs on trial as potential ICIs for HCC patients including nivolumab and atezolizumab[57], but not all HCC tumors respond well to anti-PD-1 inhibitors[73] which suggests complex PD-1/PD-L1 regulation in different HCC subsets.
In a Caucasian HCC cohort (n= 217), PD-L1 was expressed in 17% of tumors, with a positivity rate ranging from 1% to 30%. This PD-L1 expression was associated with HCC progenitor subtype with all the common markers for tumor aggressiveness including high alpha-fetoprotein (AFP) levels, satellite nodule, macrovascular and microvascular invasion, poor differentiation, and CK-19 expression[83]. Similarly, in a Chinese HCC cohort (n= 411), only 19% of tumor cells expressed PD-L1 positivity,which was correlated with high CD8+T cell densities. The high level of CD8+T cells was associated with better OS and recurrence-free survival[59]. In a different Chinese HCC cohort (n= 304), only increased PD-L2 expression was observed in 19% of tumor cells. A high proportion of PD-L1 and PD-L2 was also observed in TIICs in HCC stroma, which was correlated with higher CD8+T cells[84].
PD-L1 expression in the HCC microenvironment
PD-L1 is expressed by stromal cells in the HCC microenvironment, especially in tissue adjacent to carcinoma portal exchange and endothelial cells[52]. PD-L1-expressing inflammatory cells were identified in 76% of tumors (n= 217, Caucasian cohort), and was associated with high AFP levels, macrovascular invasion, poor differentiation,high PD-1 expression, and lymphoepithelioma-like histological subtype of HCC[83].Positive PD-L1 expression was also identified on sinusoidal lining cells (mostly Kupffer cells), endothelial cells, and ICs in adjacent non-HCC parenchyma and noncirrhotic liver (n= 68); with most PD-L1 positive cells identified as ICs[85]. CD8+CD68+Foxp3+ICs were associated with HCC especially in the invasive margin, while CD8+cells were correlated with PD-L1 positive cells[85]. Similarly, high expression of PD-1 and PD-L1 in both tumor interior and invasive margin were correlated with high densities of CD3+and CD8+T cells; which was associated with a low rate of occurrence and prolonged RFS (relapse-free survival)[86].
In a Chinese HCC cohort (n= 90), high PD-L1 positive expression (31%) was found in HCC peritumoral tissues, which was related to more vascular invasion, lower albumin level, and worse OS[63]. Another study with a bigger HCC cohort (n= 304)also found high PD-L1 expression in immune stroma (which was identified as mostly macrophages). This high PD-L1 expression was correlated with CD8+ T cells infiltration, but interestingly resulted in poorer OS and DFS (disease-free survival) outcome[84]. The difference between CD8+T cells infiltration and disease outcome in CaucasianvsChinese cohorts may be attributed to TGF-β expression. Increased TGF-β and TGF-β+Tregs were identified in the peripheral blood of HCC patients (n= 100),and patients with high TGF-β and TGF-β+Tregs had a lower OS rate. TGF-β may promote IL-6 production thereby promoting tumor growth and proliferation[87]. In addition, high TGF-β level also reduced IFN-γ secretion by CD8+T cells[88]. Lowering TGF-β level in tumor cells with high CD8+T cells seemed to improve the outcome of the disease[59].
PD-L1 expression in infiltrating ICs
High PD-L1 positivity rate in various cancers was identified in 36% of TIICs based on a meta-analysis study[89], and this high PD-L1 positivity showed a good correlation with a lower risk of death and better cancer survival. Increased PD-L1 level in TIICs was also found in post-sorafenib HCC tissues compared to pre-sorafenib tissues, and these TIICs were mostly identified as CD68+macrophages[90]. Macrophage activation in HCC has been attributed to hypoxia in the tumor microenvironment. Moreover,treatment with sorafenib has been shown to induce hypoxia in the tumor microenvironment, further inducing activation of macrophages and other ICs[64]. In addition, M1 macrophages (CD68+HLA-DR+) in HCCs can induce PD-L1 expression through IL-1β signaling[91].
TAMs were regulated by PD-L1 and osteopontin (OPN) in HCCs, and high OPN level was associated with TAMs infiltration in HCC tumor cells. High OPN expression in HCCs provides alternative activation of macrophages and facilitates the chemotactic migration of macrophages. In addition, OPN also upregulated PD-L1 expression in HCCviaCSF-1 (colony stimulating factor 1)-CSF-1R (colony stimulating factor receptor 1) pathway activation in macrophages[92]. TAMs trafficking in HCCs was blocked by inhibiting CSF-1/CSF-1R leading to enhancement of ICI efficacy in HCC treatment[92].
PD-L1 expression in HBV-related HCCs may affect the activation of follicular helper T (Tfh) cells leading to impaired B cell antibody responses. Increased PD-L1 in this cohort is accompanied by reduced expression of ICOS, secretion of IL-10 and IL-2, and Tfh proliferation. The PD-L1 effect on Tfh cells increased gradually through different HCC stages, with Tfh cells from stage III patients showing a lower effectiveness in inducing naïve B cells differentiation into plasmablasts. PD-1 blockade only partially rescued Tfh functions in HCC stage I and II, but not in stage III HCC. On the other hand, treatment with recombinant PD-L1 strongly suppressed Tfh functions in all HCC stages[93]. This study clearly defined the PD-1/PD-L1 upregulation effect on Tfh cells exhaustion in HCCs.
A novel subset of protumorigenic PD-1+B cells was identified in HCCs. These B cells expressed CD5hiCD24-/+CD27hi/+CD38dim, different to the usual CD24hiCD38hiphenotype of the peripheral regulatory B cells[94]. Upregulation of TLR4-mediated BCL-6 induced this B cell subset, while STAT-6 phosphorylation by IL-4 abolished them. This B cell subset interacted with PD-1, suppressed tumor-specific T-cell immunity, and promoted cancer growthviathe IL-10 pathway[94].
NK cells which play an important role in tumor immunosurveillance, were found in low abundance in HCC tissues compared to the adjacent normal liver tissues.However, the abundance of NK cells in HCCs was associated with various immune checkpoint proteins including PD-1, PD-L1, KLRD-1, CTLA-4, and CD86. Higher abundance of NK cells resulted in a better response to sorafenib and OS of HCC patients[95].
PD-L1 expression in HCC can also be induced by monocytes. Monocytes may greatly enhance the glycolysis process at the HCC peritumoral region, inducing PD-L1 expression in these cells but attenuating cytotoxic T lymphocyte responses in HCC tumor tissues[96]. This increased glycolysis rate was enabled by the upregulation of PFKFB-3, a glycolytic enzyme, in tumor-associated monocytes by tumor-derived soluble factors such as hyaluronan fragments. Increased PFKFB-3+CD68+cell infiltration in peritumoral HCC tissues was negatively correlated with OS[96]. In addition,PFKFB-3 also induced direct PD-L1 expression by activating the NF-κβ pathway[96]. The increase in PD-L1 and PD-L2 on monocytes (CD14+) in HCC patients was also associated with a poor prognosis[97].
SOLUBLE PD-1 AND PD-L1
Apart from its membrane-bound forms, PD-1 and PD-L1 can exist in soluble forms,sPD-1 and sPD-L1, respectively[98]. sPD-1 resulted from alternative splicing of the transmembrane domain exon 3 from the PD-1 gene. The sPD-1 level was found to be increased following PBMCs activation with anti-CD3+and anti-CD28 monoclonal antibodies, in parallel with an increased level of full-length PD-1. This observation suggests an interplay between sPD-1 and PD-1 in the maintenance of peripheral selftolerance and prevention of autoimmunity[99]. sPD-L1 possibly resulted from cleavage of membrane PD-L1 (mPD-L1) by matrix metalloproteinases; however, it still retained the IgV ligand-binding domain required for PD-1 interaction and subsequent inhibition of T cell activation[100,101]. sPD-L1 is mainly produced by myeloid-derived cells such as monocytes, macrophages, and DCs, but has also been found in several human cancer cell lines[100]. Increased sPD-L1 levels in blood were associated with metastasis and poor prognosis in breast cancer, diffuse large B cell lymphoma, and clear cell renal cell carcinoma (ccRCC)[100,101]. High sPD-L1 level was also associated with increased mortality risks in ccRCC and HCC[101,102].
The impact of sPD-1 levels on long-term dynamics of HBV load and HCC risk in 2903 Chinese HBV patients showed that sPD-1 levels were associated with higher viral load for more than four consecutive years and increased risk of HCC, especially in male patients. The high levels of sPD-1 and HBV load was also associated with a 6-fold increase in HCC risk[103], showing an association between high sPD-1 level and HCC development. Similarly, sPD-L1 levels were positively correlated with stages of liver cirrhosis and HCC in a cohort of 215 Caucasian HCC patients. Patients with a high sPD-L1 level had an increased risk of mortality, while those with low sPD-L1 had a better prognosis[102].
It is interesting to note that a recent study on soluble PD-1 and PD-L1 in HCC patients, concluded that sPD-1 and sPD-L1 were independent prognostic biomarkers with opposite effects in HCC, with sPD-1 level a favorable prognostic factor for HCC patients[98]. They found detectable sPD-1 in all HCC patients’ sera (n= 120), while sPD-L1 level was only detectable in two-thirds of the patients, although sPD-L1 level seems to be positively correlated with sPD-1 level[98]. Furthermore, there was no association found between sPD-L1/sPD-1 level and intratumoral expression of PD-L1 level or numbers of CD4+and CD8+TILs[98]. Several studies have reported that sPD-1 may suppress the PD-1/PD-L1 pathway leading to restored T cell function and enhanced antitumor immunity[104,105], which may explain the results by Changet al[98], which showed a favorable sPD-1 level with HCC progression. More studies are needed to elucidate the role of sPD-1 and sPD-L1 in HCC development.
BIOMARKERS FOR ANTI-PD-1/PD-L1 ICIS
Since the benefit of ICIs targeting the PD-1/PD-L1 is restricted to a subset of patients,predictive biomarkers are essential for patient selection[106]. Several putative markers have been proposed with predictive potential, but the strongest proven marker to date is the expression of PD-L1 assessed by immunohistochemistry (IHC)[106].
Further study on tumor samples from dose-escalation and dose-expansion phases of the anti-PD-1 trial, the CheckMate 040[107], showed that tumoral expression of PD-1 and PD-L1 was associated with improved OS. The percentage of PD-1+cells was higher in responders or partial responders compared to patients with stable or progressive disease, where tumor PD-L1 expression ≥ 1% was associated with improved OS. Gene analysis by RNA-seq also showed that the inflammatory signature consisting of CD274 (PD-L1), CD8A, LAG-3, and STAT-1 was associated with improved patient survival and response to anti-PD-1[108]. In a study of cytokineinduced killer (CIK) cell immunotherapy, patients with higher PD-L1 expression were those who exhibited long-term survival benefit post CIK[76].
Higher response to anti-PD-1 was observed among patients with a high intratumoral CD38+cell proportion in the tumor microenvironment[109]. Previously it was demonstrated that TILs expressing activation marker CD38 in the tumor was correlated with patient survival, indicating that enhanced local immune activation contributes to a better prognosis for patients with HCC[110].
In line with the above studies, anti-PD-1 treatment failure was associated with the upregulation of alternative immune checkpoints that limit the antitumoral immune response[111]. Adaptive resistance to anti-PD-1 treatment was shown to correlate with the upregulation of indoleamine 2,3-dioxygenase (IDO) and alternative checkpoints for TIM-3[112,113]. In their study, Koyamaet al[113] sorted T cells and tumor cells by mRNA sequencing and flow cytometry of anti-PD-1-resistant cellsvsuntreated tumors. TIM-3, LAG-3, and CTLA-4 were expressed at higher levels in PD-1-resistant cells, but only TIM-3 showed a significant increase. In a mouse model, resistance to PD-1 blockade was overcome by the addition of TIM-3 antibody[113]. Higher serum levels of TIM-3 have been correlated with advanced HCC stage, poor prognosis, and patient’s response to TACE[114,115]. Univariate logistic regression showed that higher serum TIM-3 values were associated with a higher probability of serum PD-L1 detection[115], which might be related to the simultaneous activation of both immune checkpoints in advanced HCCs.
Systematic interrogation of TILs is key to the development of immunotherapy and the prediction of their clinical responses in cancers[116]. In the study by Zhenget al[116], single-cell RNA-sequencing analyses of > 5,000 single T cells isolated from HCC patients showed that specific subsets such as exhausted CD8+T cells and Tregs, with high expression of PDCD1 were preferentially enriched and potentially clonally expanded. Layilin (LAYN) was upregulated on activated CD8+T cells and Tregs.In vitro, LAYN overexpression in primary CD8+T cells resulted in inhibition of IFN-γ production, suggesting a regulatory function of LAYN[116].
Genomic mutations including single nucleotide polymorphisms (SNPs) have been associated with HCC risk, including predisposition to risk factors, the severity of liver disease, malignant transformation, and tumor progression[117]. Tumor mutational burden (TMB), defined as the total number of somatic mutations per megabase or the nonsynonymous mutations in tumor tissues, including replacement and insertiondeletion mutations[118], has been associated with the success of ICIs therapy. In a meta-analysis report of 2,661 patients from 8 trials (mostly of lung cancer), patients with high TMB showed significant benefits from PD-1/PD-L1 inhibition compared to patients with low TMB[118]. The significance of TMB as a biomarker in anti-PD-1/PDL1 treatment was also reported in several independent studies[119-121].
However, this information is still lacking for HCC. The evaluation of the frequency of genomic biomarkers including the TMB in 755 patients of advanced HCC showed no significant genomic or TMB differences between responsive patients and those with progressive or stable disease. Furthermore, PD-L1 positivity was not associated with high TMB, where several patients with high positive PD-L1 were also TMB low (2-5 mutations/Mb)[122]. These data were confirmed by more recent studies showing that TMB could not predict OS and patient’s responsiveness to anti-PD-1[123-125]. Besides,HCC had low levels of microsatellite instability, a phenotype due to accumulated mutations resulting from a defect in mismatch repair[126].
PD-1/PD-L1 GENETIC VARIATIONS
PD-1 polymorphisms
PD-1 is encoded by the PDCD1 gene located on chromosome 2 which contains five exons, while PD-L1 is encoded by the CD274 gene located on chromosome 9 which contains seven exons[36]. Various genetic aberrations that can affect PD-1/PD-L1 gene expression have been identified, including SNPs, copy number alterations (CNAs),amplifications, deletions, mutations, and spliced variants (Table 1).
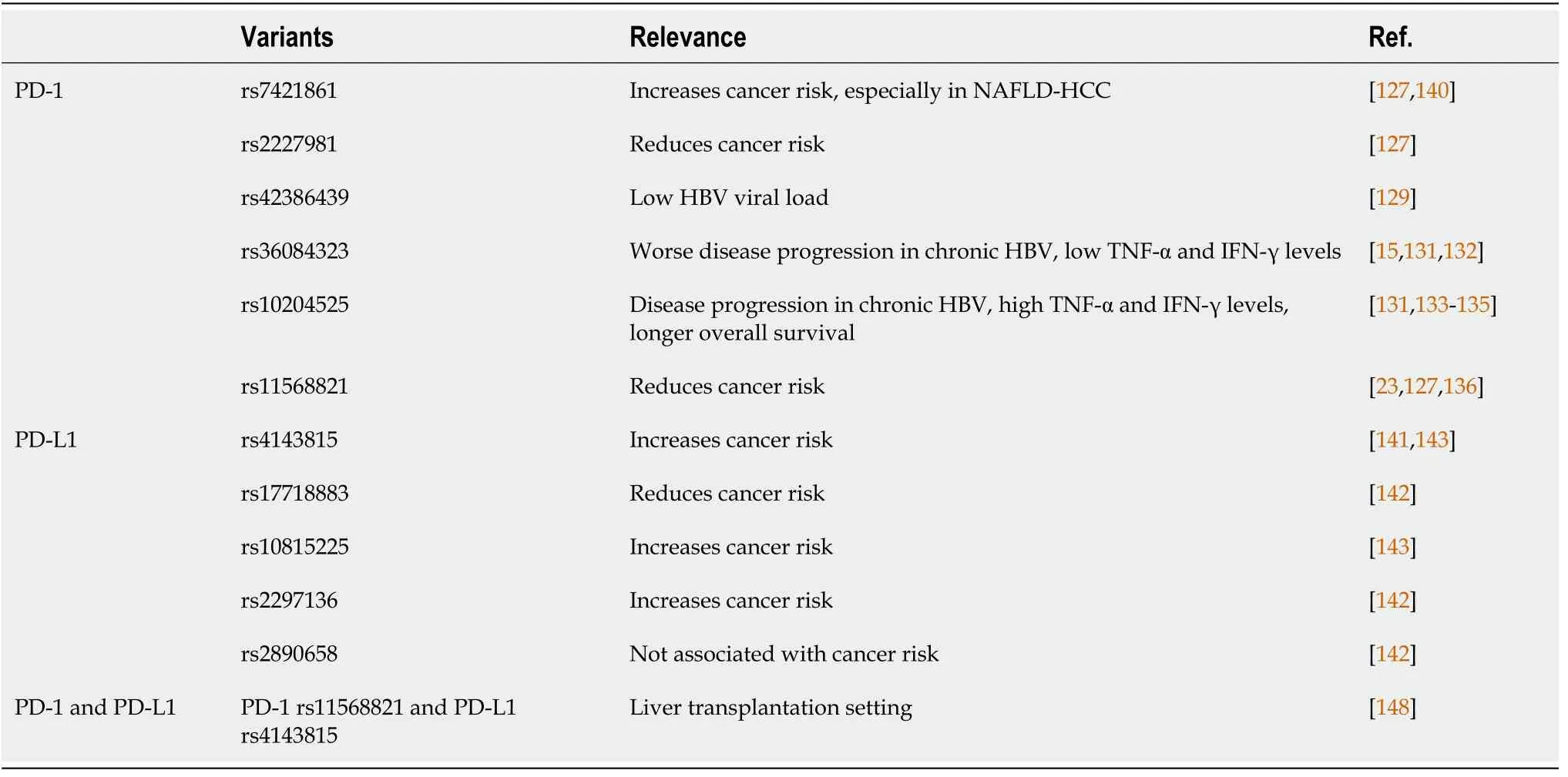
Table 1 Programmed death-1/programmed death ligand 1 nucleotide variations and their clinical relevance in liver disease
Several PD-1 gene polymorphisms have been identified, including six of the most studied PD-1 SNPs concerning various cancers. One is located in-frame rs2227982(C>T), resulting in amino acid mutation from alanine to valine (A215V), two located upstream in the promoter region rs2227981 (C>T) and rs36084323 (G>A), two in the intron region rs7421861 (T>C) and rs11568821 (G>A), and one in the 3’UTR region[rs10204525 (G>A)][15]. A meta-analysis of these SNPs on various human cancers showed that rs7421861 polymorphism was associated with increased risk of developing cancer, while rs2227981 and rs11568821 polymorphisms were associated with overall reduced cancer risk[127]. The cancer-protective effect of PD-1 genotype can be seen in both the Asian and Caucasian populations[128].
Earlier studies have indicated the association of several PD-1 gene polymorphisms with chronic HBV liver disease progression in Chinese patients. PD-1 rs42386439 T allele, located in intron 4, which acts as a negative cis-element for gene transcription,was significantly associated with lower HBV viral load[129]. PD-1 rs36084323 SNP was associated with liver disease progression to cirrhosis and HCC in chronic HBV patients[130]. In addition, rs36084323 AA genotype in chronic HBV patients was associated with overall lower TNF-α and IFN-γ levels[131]. Higher PD-1 rs36084323 AA genotype but lower rs2227981 TT genotype frequencies were also found in chronic HBV patients compared to the spontaneously recovered control group[132]. PD-1 rs36084323 polymorphism, located in the PD-1 promoter region, may interrupt PD-1 gene activation and transcription, thus affecting T cell activation and function and altered cytokine secretion, possibly resulting in a worse prognosis of liver diseases including cancer development[15,131].
PD-1 rs10204525 polymorphism, located in the 3’UTR, was also associated with progression of HBV-related liver disease[133]. PD-1 rs10204525 GG genotype HBV patients had a higher level of TNF-α, possibly conferring a strong inhibitory effect on PD-1 function and subsequent T-cell activation[131]. A functional study on lymphocytes from chronic HBV patients showed that allele G of rs10204525 binds specifically to miR-4717 leading to decreased PD-1 expression but increased TNF-α and IFN-γ levels. The same effect was not seen in allele A of rs10204525[45]. Indeed, PD-1 rs10204525 AA genotype was associated with liver cirrhosis in chronic HBV patients,and in combination with TIM-3 (T-cell immunoglobulin mucin-3) rs10053538 GT or TT genotypes were more frequently found in HBV-associated HCC patients[134]. PD-1 rs10204525 GG+AG genotypes were also significantly associated with longer OS in HBV-associated HCC patients receiving various treatments including surgical treatment, TACE, and other supportive treatments[135].
PD-1 rs11568821 variants, an intronic SNPs, affect PD-1 mRNA level by changing its binding affinity to RUNX (a PD-1 transcriptional factor), resulting in impaired PD-1 inhibitory effect and subsequent positive regulation of cytotoxic T lymphocyte activity[15,23]. As such, PD-1 rs11568821 GG genotype has been associated with decreased cancer risk[23,136]. However, a 2012 Turkish study found no significant differences in PD-1 rs11568821 genotype between HCC cases and control subjects[137]. A study performed in 2015 also found no significant differences in the genotype distributions of PD-1 rs11568821 and rs41386439 in chronic HBV patients compared to the spontaneously recovered control group[138], indicating the possibility of lack of association between PD-1 gene polymorphisms and HBV infection susceptibility and HBV-related HCC progression in Turkish patients. These findings were confirmed in a more recent study investigating three PD-1 gene polymorphisms and HCC progression; again no significant distribution of rs36084323, rs2227981, and rs10204525 genotypes were observed in HCC cases in Turkish patients[139].
PD-1 rs7421861 SNP, located in intron 1, may disrupt the putative alternative splice site and promote full-length transcript expression instead[15,140]. An earlier study showed no association between rs7421861 variants and cancer risks[136]. However, a more recent study in NAFLD-HCC European cohorts revealed that PD-1 rs7421861 allele A was significantly associated with HCC, independent of age, sex, cirrhosis, and diabetes. Furthermore, allele A of rs10204525 in this cohort was also associated with increased risk of NAFLD-HCC, especially in female patients[140]. These findings revealed the association between PD-1 gene polymorphisms not only in HBV-related HCC but also in NAFLD-HCC.
PD-L1 polymorphisms
Similar to PD-1 polymorphisms, several PD-L1 polymorphisms, rs4143815 (C>G),rs2890658 (A>C), rs2297136 (C>T), rs17718883 (C>G), and rs10815225 (G>C), have been studied in association with the development of various cancers. A meta-analysis showed that PD-L1 rs4143815 polymorphism was associated with protection against various cancers[127]. However, different studies have shown that carriers of PD-L1 rs4143815 GG genotype have a higher risk of developing gastric adenocarcinoma[44]and HCC[141].
A 2018 study examining several PD-L1 variants in 225 Chinese HCC patients confirmed that PD-L1 rs4143815 GG and rs2297136 TT genotypes were associated with increased HCC risks. On the other hand, PD-L1 rs17718883 CG+GG genotypes reduced the risk of HCC occurrence, while rs2890658 SNPs were not associated with HCC risks[142]. PD-L1 rs4143815 polymorphisms, located in the 3’UTR, resulted in elevated PD-L1 protein expressionviadisruption of miR-570 binding to PD-L1 mRNA[44]. This polymorphism was found in disequilibrium with PD-L1 rs10815225 polymorphism in the PD-L1 promoter region, which serves as a binding site for Sp1 transcription factor (SP1). The G allele of rs10815225 bound more effectively to SP1 resulting in an increased level of PD-L1 mRNA level. PD-L1 rs10815225 GG genotype was associated with increased risk of gastric cancer, and the haplotype of rs10815225 and rs4143815 polymorphisms were found to greatly increase gastric cancer risk[143].
PD-1 and PD-L1 polymorphisms interaction
Aside from the association of singular PD-1 and/or PD-L1 polymorphisms and cancer progression, the interaction between multiple SNPs of PD-1/PD-L1 with various genes has also been shown to be associated with HBV infection and related cancer development. A combination of PD-1 rs10204525 GG and rs2227982 CC genotypes in Chinese patients has been shown to result in better protection from HBV infection and lower HBV viral load in asymptomatic carriers[144]. On the other hand, the interactions between PD-1 rs41386349 and rs6710479 with TIM-2 rs246871 variant were shown to affect susceptibility to chronic HBV infection and may influence later hepatocarcinogenesis[145]. Furthermore, PD-1 rs10204525 AA genotype and TIM-3 rs10053538 GT or TT genotypes were more frequently found in HBV-associated HCC patients[134].
Similarly, PD-1 rs11568821 synergy with CTLA-4 49AG:CT60 A:A haplotype has been associated with an increased risk of primary biliary cancer (PBC)[146].Interactions between PD-L1 rs10815225 and PD-1 rs7421861 polymorphisms were also associated with the development and outcome of ccRCC[147]. No such observation has been reported so far for HCC, although the interaction between PD-L1 rs4143815 and PD-1 rs11568821 variants was found to be important in the liver transplantation setting. PD-L1 rs4143815 was associated with different PD-L1 expression on donor hepatic DCs upon IFN-γ stimulation, and PD-1 rs11568821 A allele recipients receiving donors from PD-L1 rs4143815 GG genotypes had a higher risk for late acute rejection after liver transplantation[148].
PD-L1 genetic alterations
Aberrant PD-L1 expression may be caused by PD-L1 genetic alterations affecting the PD-L1 locus. PD-L1 CNAs affecting either the focal regions, chromosome 9p24.1, or the whole chromosome 9 have been identified in 22 major cancer types, resulting in changes in PD-L1 mRNA expression[74]. These PD-L1 variants significantly affected PD-L1 expression, and a higher PD-L1 expression was observed in cancer patients with altered PD-L1 variants, with PD-L1 gene fusion and amplification showing the highest increase in PD-L1 expression[149]. PD-L1 copy number gains and deletions were associated with higher mutational loads, while PD-L1 amplifications and deletions of core regions were associated with a more dismal cancer prognosis[150].PD-L1 deletions were more frequent in solid tumors, especially in melanoma and NSCLC where more than half of the tumors had PD-L1 deletions[14,150].
In liver cancer, PD-L1 copy number gains were associated with increased JAK-2 mRNA expression[150]. It is interesting to note that JAK-2 and PD-L1 encoding genes are both in chromosome 9p, with both having high alternation rates. JAK-2 amplification and mutation which increased JAK-2 and its downstream STAT effectors expression has also been shown to upregulate PD-L1 expression[41,51]. A 2018 study on Chinese HCC patients revealed a significant proportion of chromosome 9p24.1 polysomy (16%-31%) and amplification (7%-15%). Furthermore, these PD-L1 genetic alterations were significantly associated with upregulation of both PD-L1 and PD-L2 expression, high infiltration of PD-1+ICs, and overall poor cancer survival[151].
PD-1/PD-L1 spliced and soluble variants
A high expression level of PD-L1 truncated form was first identified in a head and neck squamous cell carcinoma (HNSCC), as a result of the human papillomavirus integration into the PD-L1 locus upstream of the transmembrane domain-encoding region[152]. A follow-up study on 33 cancer types and human cancer cell lines identified additional PD-L1 truncated forms in 20 cancers and human cancer cell lines,characterized by exon 4 enrichment. This truncated PD-L1 was preferentially secreted but still maintained its binding ability to PD-1 and served as a negative regulator for T cell activation by inhibiting IL-2 and IFN-γ secretion[153].
A different secreted splice variant of PD-L1 (secPD-L1) has been identified in various tumor types and malignant cell lines. This secPD-L1 contains the first exons of PD-L1 but lacks exon 5 and cannot splice into the transmembrane domain. However,due to its 18 amino acids tail containing tyrosine, this variant can homodimerize and inhibit T-cell proliferation and IFN-γ production, retaining its immunosuppressive effect[154]. PD-L1 secreted (secPD-L1) splice variants lacking the transmembrane domain were identified in NSCLC patients. These secPD-L1 variants mediated resistance to PD-L1 blockade therapy by acting as a decoy to PD-L1 antibody, which was found to be constrained by treatment with anti-PD-1 antibody[155].
HCC CELLULAR HETEROGENEITY
HCC molecular classification
HCC is a vast heterogeneous malignancy, either within an individual (intratumoral heterogeneity) or among subjects (intertumoral heterogeneity). Besides its various underlying etiologies, long-term development affects its different profiles and distinct progression of the disease. In the ‘-omics’ era with abundant studies of global gene screening and genetic array, HCC classifications have shifted from histology[156] to molecular-typing-based. HCC (sub)types can arise from dysregulations of various oncogenic pathways and/or different cells of origin[157]. Starting in the 2000s, several studies demonstrated that HCCs could be categorized into specific subclasses based on their distinct molecular signatures (Table 2).
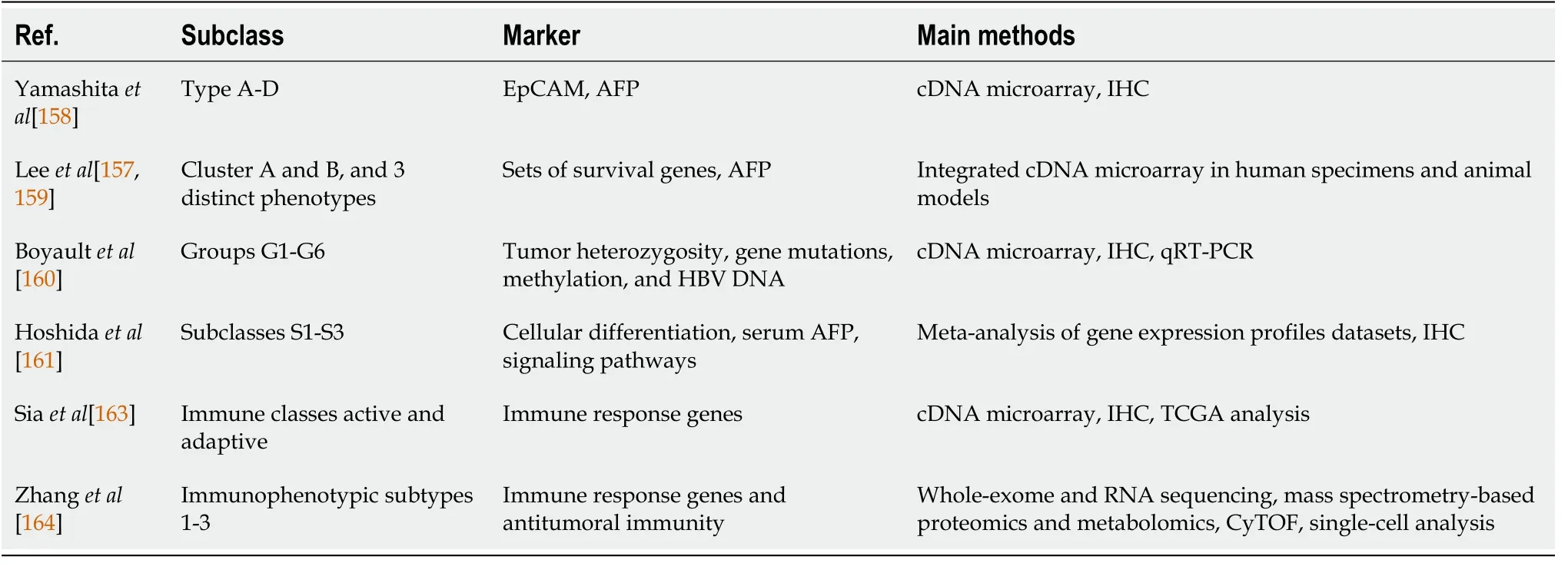
Table 2 Major molecular classifications of hepatocellular carcinoma
HCC classification based on progenitor cells was identified using the CSC marker EpCAMviacDNA microarray analysis and IHC analysis. EpCAM+HCC displayed the features of hepatic progenitor cell markers (e.g., CK-19, c-Kit, EpCAM, and activated Wnt/β-catenin), whereas EpCAM-HCC displayed genes with features of mature hepatocytes. The two groups were then classified into four subgroups with a high or low level of AFP. Kaplan-Meier analysis showed that EpCAM+AFP+(type B) and EpCAM−AFP+(type C) HCCs were correlated with poor prognosis, whereas EpCAM−AFP−(type D) HCCs was correlated with an intermediate prognosis. Interestingly,EpCAM+AFP−(type A) HCCs was correlated with a good prognosis[158].
In 2004, using cDNA microarray, the Thorgeirsson group classified HCCs into clusters A and B. Cluster A, in combination with low serum AFP, was associated with low survival[159]. Using integrated gene expression data between human specimens and animal models, the group further classified HCC into three distinct biological phenotypes with significant differences in clinical outcome. The hepatoblast (HB)subtype belonged to cluster A, while the hepatocyte (HC) subtype belonged to both cluster A and B. Differential expression of around 1500 genes showed exclusive differences between HB and HC subtypes, especially in the pathway of the JUN-FOS heterodimers–AP-1 complex. Individuals with HCC who shared a gene expression pattern with HB subtype had a poor prognosis[157].
Also using global transcriptome analysis, Boyaultet al[160] classified HCCs into subgroups G1 to G6, each associated with specific clinical and genetic characteristics,especially tumor heterozygosity, gene mutations, promoter DNA methylation, and HBV DNA copy number. The subgroup G1 and G2 were related to activation of the AKT pathway, while G5 and G6 to β-catenin mutations leading to Wnt pathway activation. In brief, the characterization of G1 was related to HBV low copy number and fetal liver gene expression, G2 was associated with HBV high copy number,PIK3CA, and TP53 mutation, and G3 was associated with TP53 mutation and overexpression of cell-cycle genes. Subgroup G4 heterogeneously comprised TCF-1 mutated hepatocellular adenomas and carcinomas. Subgroup G5 was associated with stress and immune response, and G6 with amino acid metabolism and satellite nodules[160].
In 2009, Hoshidaet al[161] performed a meta-analysis of gene expression profiles from 8 independent datasets, validated by immunohistostaining of clinical specimens.Here, they classified HCC into 3 robust subclasses S1 to S3 which were correlated with clinical parameters. Subclass S1 was characterized by stromal cells with TGF-β activation, S2 by stem-angiogenic cells with MYC and AKT activation, and S3 by mature hepatocyte differentiation. The subclass S2 with EpCAM positive cells,categorized as stem cells-like and hepatoblast-like HCC, was also highly aggressive[161], as noted in previous studies[157,159]. A more recent study from the group showed that HCC S1 and S2 were found in various established cell lines, thus indicating the appropriate use ofin vitromodels to evaluate the effectiveness of subtype-specific drug response. For example, (+)-JQ1, an anti-MYC compound, was highly sensitive in S2 cell lines HepG2 and Huh7[162].
From an immunological point of view, several studies have categorized HCC based on the tumor microenvironmentviathe infiltration of ICs. By using gene expression profiles from the tumor, stromal, and ICs, followed by immunohistochemical analysis,Siaet al[163] identified two robust HCC immune classes. HCC data in The Cancer Genome Atlas (TCGA) were analyzed to correlate the ICs gene expression profiles with chromosomal aberrations and mutations. The active immune response subtype(approximately 65%) was characterized by overexpression of adaptive immune response genes, while the exhausted immune response subtype (approximately 35%)was characterized by the presence of immunosuppressive signals (e.g., TGF-β, M2 macrophages)[163]. This finding indicated the susceptibility of HCC upon immune modulation therapy against T cells.
A recent paper by Zhanget al[164] further confirmed the heterogeneity of the HCC microenvironment in a complex and integrated multiomics analysis. The immune status of the HCC microenvironment was relatively less heterogeneous, thus rendering the significance of HCC immunophenotypic classification[164]. Using whole-exome sequencing, RNA sequencing, mass spectrometry-based proteomics and metabolomics, CyTOF, and single-cell analysis, the authors classified HCC based on its immunophenotypic subtypes. By clustering the ICs in the HCC microenvironment,they identified three distinctive novel HCC subtypes 1 to 3 with immunocompetent,immunodeficient, and immunosuppressive features. Subtype 1 was characterized by relatively normal T cell infiltration levels but fewer infiltrating B cells, whereas subtype 2 had reduced infiltration of lymphocytes but high frequencies of DCs and NK cells; subtype 3 had high frequencies of Treg cells, Breg cells, and M2-polarised macrophages. HCC samples with subtype S3 showed significant upregulation of immunosuppressive molecules including PD-1, PD-L1, TIM-3, and CTLA-4. These three HCC subtypes were also associated with the clinical situations in patients.Patients in subtype 1 with competent antitumoral immunity showed good prognosis[164].
HCC cancer stem cells
The population of cancer stem cells (CSCs), also known as tumor-initiating cells (TICs)or side population (SP), has been recognized as one if not the most important cells in cancer. They are responsible for the initiation and the maintenance of various types of cancer, while also contributing to tumor resistance during treatment.
In HCC, various protein markers, including the CD133/Prom-1, CD90/Thy-1,EpCAM, CD24, CD13/ANPEP, ABCG2/BCRP, aldehyde dehydrogenase/ALDH,CD44, and many more, have been proposed to define and to isolate the CSCs from the tumor ‘bulk’ populations[165]. Furthermore, the combinations of using two or more markers added to the variations of CSC populations. In fact, until now there is no consensus on the use of CSC markers for HCC. It is important to note that each CSC population had its distinct characteristics[166].
The origin of hepatic CSCs is various, thus further increasing the vast cellular variations within the tumor. In the beginning, it was thought that CSCs were derived exclusively from oncogenic transformation in normal stem/progenitor cells[167]. Due to the complexity and physiology of the liver, however, the origin of hepatic CSCs can be traced into multiple lineages of liver maturation. In 2013, the Thorgeirsson group provided strong direct evidence on various sources of hepatic CSCs. Upon controlled oncogenic transformation with H-ras and SV40LT, they noted that adult hepatocytes,hepatoblasts, and hepatic progenitor cells (HPC) could be oncogenically reprogrammed into hepatic CSCs. While all three lineages possessed CSC properties, they showed different tumorigenic potential with HPC-tumors having the highest[168,169].Depending on the origin of the cell on which the malignant transformation occurred, a broad range of different liver cancer phenotypes, from classic HCCs and iCCA(intrahepatic cholangiocarcinoma) to mixed HCC–iCCA lesions, was also observed[168,170].
Besides its main capacity to induce tumor, the main feature of the CSC population is their resistance to various treatments. The CSC populations (e.g., CD133, CD13,EpCAM) are highly resistant to chemotherapy and radiotherapy[171-175], and sorafenib[176-181]. On the other hand, the preferential expression of CSCs and their response to immune therapy is still unclear.
EXPRESSION OF PD-L1 IN CSC POPULATIONS
Immunotherapy is a rather new field in cancer study. It is still unclear whether the success of ICIs has any association with HCC cellular hierarchy. CSCs have been demonstrated to have a preferential role in therapy resistance, including in chemotherapy, radiotherapy, and molecular therapy, thus it is reasonable to investigate the potential relevance of CSCs against immunotherapy. CSC tumoral heterogeneity might have a close association with the intrinsic PD-L1 properties in cancer cells.
Until now, contrasting studies showed the association of PD-L1 to tumorigenesis and CSCs (Table 3). In breast and colorectal cancer, PD-L1 was positively correlated with CSC populations, regardless of their phenotypic markers. Using flow cytometry analysis, authors have shown that PD-L1 expression was higher in CSCs of both cancers compared to non-stem like cancer cells. High PD-L1 expression was noted in CSC subpopulations expressing CD44+, ALDH+, CD44hiCD24lo, EpCAM+CD90hi, and EpCAM+CD44hiCD24lo[182-184]. For example, in CSC EpCAM+CD44hiCD24lo, PD-L1 was overexpressed up to 3-fold compared to more differentiated-like cancer cells.Functionalin vitroandin vivoassays also showed higher stemness of PD-L1hias compared to PD-L1locells. Among the different pathways examined, PD-L1 expression on CSCs was partly dependent on Notch and/or PI3K/AKT pathway activation[184].
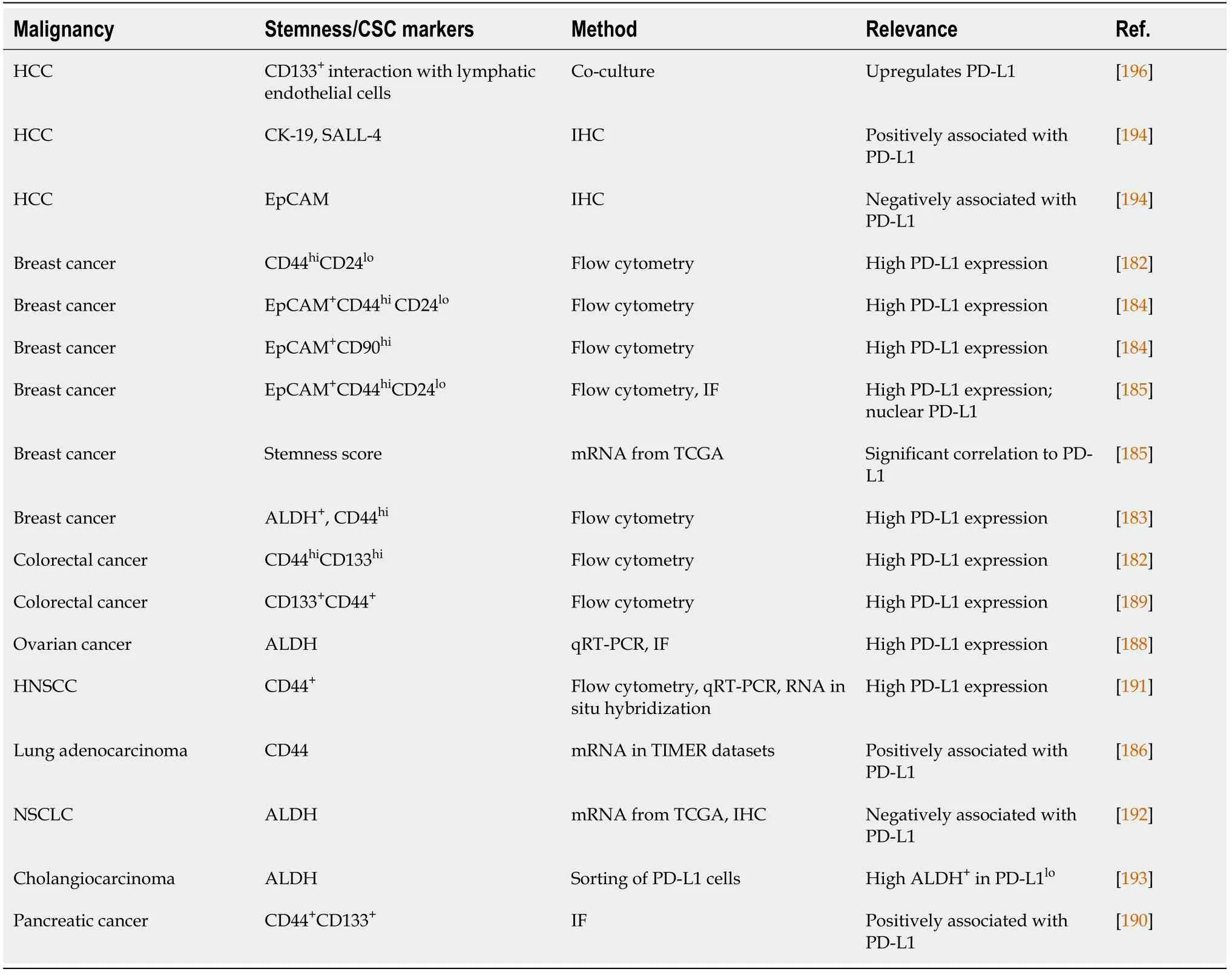
Table 3 Association between programmed death-1 expression and cancer stem cell markers
Analysis of stemness score from TCGA data of 530 breast cancer patients on PD-L1 expression and CSCs, also indicated a strong association between PD-L1 expression and stem-like cells[185]. EpCAM+CD44hiCD24loCSCs had a higher level of PD-L1 compared to their differentiated counterparts (EpCAMlo/negCD44loCD24hi). Immunofluorescence results also confirmed the higher level of PD-L1 expression in CSCs compared to the more-differentiated breast cancer cells. Interestingly, in addition to membranous PD-L1 there was also a PD-L1 nuclear fraction in CSCs. PD-L1 knockdown on the expression of stem-related molecules suggests a direct role for this molecule in CSC maintenance[185]. A positive correlation between mRNA expression of CD44 and PD-L1 was observed in lung adenocarcinoma using Tumor Immune Estimation Resource (TIMER) datasets, which was validated using IHC[186].
In gastric cancer, a CSC cell line, the NCC-S1M, overexpressed PD-L1 compared to normal gastric tissue. Furthermore, anti-PD-1 treatment suppressedin vivogrowth of CSC-like cell allografts in syngeneic mice. PD-L1 was controlled by transcriptional factor Smad-4[187]. In ovarian cancer, in bothin vitromodel andin vivomouse model,higher expression of PD-L1 was observed in CSCs (ALDH+) than in non-CSC cells[188]. In colorectal and pancreatic cancer, CD44hiCD133hiCSCs also expressed high PDL1[182,189,190]. Overexpression of PD-L1 promoted colorectal CSCs self-renewalin vitroandin vivo, and increased its chemoresistance[189]. In HNSCC, PD-L1 was also highly expressed in CD44+cells[191].
However, in NSCLC and cholangiocarcinoma, the expression of PD-L1 was inversely associated with ALDH-expressing cells[192,193]. mRNA analysis of TCGA data showed that PD-L1 expression was negatively correlated with ALDH-1 expression in adenocarcinoma, also observed by IHC[192]. In cholangiocarcinoma, PDL1locells isolated from cell lines were highly tumorigenic compared to PD-L1hicells.These cells had high ALDH activity, reduced reactive oxygen species production, and were in a dormant state of the cell cycle. Furthermore, in clinical specimens, the low expression of PD-L1 was well-correlated with poor prognosis of patients[193].
With regard to HCC, to date, information on the direct association between PD-L1 and hepatic CSCs is still very limited. Nishidaet al[194] analyzed 154 HCCs and theirnoncancerous liver tissue counterparts for the expression of PD-L1 and stemness. They showed that PD-L1 was frequently expressed in stem cell features of HCC. The expression of PD-L1 was associated with aggressive high-grade tumors. Using IHC,the presence of PD-L1 was positively associated with cytokeratin 19 (CK-19) and Sallike protein 4 (SALL-4), but not with EpCAM[194]. Previously, it was shown that SALL-4 regulated the stemness of EpCAM-positive HCC. The activation of SALL-4 enhanced CSC spheroid formation and invasion capacities and upregulated the expression of CK-19, EpCAM, and CD44 in cell lines[195].
Another study showed that HCC CSC CD133+preferentially interacts with lymphatic endothelial cells. Lymphatic endothelial cells create a CSC-microenvironment through direct contact with CSCs. Co-culture of CD133+cells with lymphatic endothelial cells stimulated IL-17A expression that further promoted the immune escape of CD133+ cells through the upregulation of PD-L1. These data showed that the tumor niche promoted the self-renewal and immune escape of CSCsviaPD-L1[196].
The presence of PD-L1 in the circulating tumor cells (CTCs) was demonstrated to be a prognostic and predictive biomarker for HCC patients. CTCs expressed various phenotypic profiles such as EMT and stem cell markers. Phenotype profiling of HCC CTCs in patients was performed by CTCs isolation and enrichment with an HCCspecific antibody cocktail including CSC marker EpCAM, and stained with antibodies against pan-cytokeratin (CK), CD45, and PD-L1, together with DAPI. Survival analysis showed that patients with PD-L1+CTCs (DAPI+CK+PD-L1+CD45-) had significantly worse OS compared to patients without PD-L1+CTCs (DAPI+CK+PD-L1-CD45-)[197].
MICROENVIRONMENT, CSC, AND PD-L1
How the PD-L1 pathway is involved in the tumorigenicity of hepatic CSCs is still unclear. PD-L1 is known to be transcriptionally upregulated upon EMT in the cancer microenvironment. By overexpressing and knocking-down the PD-L1 in sorafenibresistant cells, PD-L1 expression promoted EMT and cellular migratory and invasive abilitiesviathe PI3K/Akt pathway[58].
The tumor microenvironment (Figure 2) is crucial for the self-renewal and maintenance of hepatic stem cells, which may lead to the development of HCC[196].Immunologic mechanisms such as chronic inflammation due to chronic viral hepatitis or metabolic diseases play a crucial role in the initiation, development, and progression of HCC. Thus, it is important to understand the underlying mechanisms shaping the unique HCC tumor microenvironment[198].
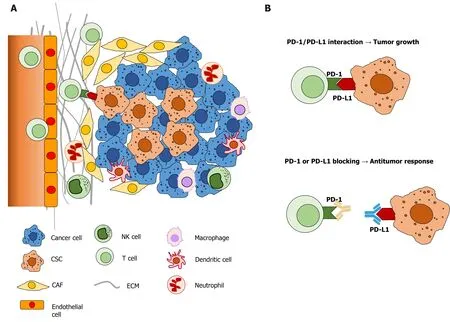
Figure 2 The complexity of hepatocellular carcinoma and its microenvironment. A: Hepatocellular carcinoma and its tumor microenvironment are composed of various cell populations, including differentiated cancer cells, cancer stem cells, cancer associated fibroblasts, immune and endothelial cells; B:Programmed death-1 and programmed death ligand 1 interaction and inhibition by neutralizing antibodies in tumor growth. PD-1: Programmed death-1; PD-L1:Programmed death ligand 1; CSC: Cancer stem cells; CAF: Cancer associated fibroblast; ECM: Extracellular matrix; NK: Natural killer.
Many of the above studies showed that PD-L1 was preferably expressed in CSCs with the phenotypic marker CD44[182-185,191]. CD44 was previously shown to function as an independent marker of hepatic CSCs[199-203]. CD44 expression was also associated with the EMT phenotype in HCC cell lines, and knocking down CD44 resulted in the switch back to the mesenchymal-epithelial-transition (MET)[199]. CD44 is a multidomain, transmembrane platform, a major adhesion molecule of the extracellular matrix. It is a signaling molecule that connects the microenvironment with growth factor and cytokine signals and regulates a variety of gene expression levels related to cell-matrix adhesion, cell migration, proliferation, differentiation, and survival[204,205]. Its ligation to growth factors was demonstrated to be able to induce partial or full EMT[206]. Our previous study demonstrated that the inhibition of hyaluronic acid in the HCC microenvironment resulted in the decreased expression of CD44 in a transgenic mouse model and HCC cell lines[207].
CSC CD44+cells were less immunogenic than CD44-cells when cultured with autologous CD8+TILs. IFN-γ treatment preferentially induced even further PD-L1 expression on CD44+cells and was associated with enhanced IFN-γ receptor expression and phosphorylation of STAT-1. Long-lived CD44+TICs can selectively evade host immune responses[191]. A subsequent study showed that EMT preferably enriched the PD-L1 in CSCs compared to the general cancer population through the EMT/β-catenin/STT-3/PD-L1 signaling axis. EMT transcriptionally induced Nglycosyltransferase of STT-3 through β-catenin, and subsequent STT-3-dependent PDL1 N-glycosylation stabilizes and upregulates PD-L1[208].
Several studies also hinted at the central role of P13K/AKT and mTOR molecular pathways in the biology of PD-L1-expressing CSCs[184,185,209]. The inhibition of STAT-1 and STAT-3, AKT downstream transcription factors, downregulated PD-L1 expression[210]. In HCC, genetic alterations involved in the PI3K/AKT pathway were significantly associated with PD-L1 positivity whereas mutations in the β-catenin pathway were inversely correlated with PD-L1 in HCC. Comparisons in the TCGA cohort showed that mutations in the PI3K/AKT pathway could positively affect the expression of PD-L1, while mutations in the β-catenin pathway were related to the absence of PD-L1 expression[194].
CONCLUSION
HCC is an immunologic cancer; therefore, immunotherapy is one of the potential treatment methods. On the other hand, HCC is vastly heterogeneous which might hamper the efficacy of therapy, especially in patients who cannot receive surgical interventions. Genetic variations in PD-1 and PD-L1 genes have been associated with the progression of liver disease. Even though its relevance in anti-PD-1/PD-L1 therapy is still lacking, these variations could be useful in determining patient acceptance of HCC treatment, also for sorafenib.
Targeting immune checkpoint ligands present in tumor cells and the microenvironment (e.g., PD-L1) can be an interesting approach. Anti-PD-L1 inhibits both the cell’s constitutive expression and inductive stimulation caused by binding of the ligand to the immunomodulatory molecule (e.g., PD-1/PD-L1). It can be given in combination with other molecular targeted therapies, which increase the ‘targeting’ of the therapy,as had been demonstrated in the co-treatment approach of atezolizumab (anti-PD-L1)plus bevacizumab (targeting VEGF)[28].
High expression of PD-L1 protein in tumoral cells and/or tumor microenvironment is indicated as a strong biomarker for the success of the ICIs against PD-1/PD-L1.Several important findings, however, need to be elucidated to measure the real efficacy of the treatment. Technical variations among laboratories may influence the results. For example, the use of the correct antibody for PD-L1 detection. A recent study showed that PD-L1 was expressed in inflammatory cells within the HCC tissue and cirrhotic parenchyma, but not in neoplastic cells[211]. Upon comparison of several anti-PD-L1 clones, the authors did not find PD-L1 immunoreactivity in both neoplastic and normal hepatocytes[211]. Different clones of anti-PD-L1 give different results thus affecting its efficiency and detection results[211-214].
Another important matter that must be considered is the use of the proper marker to characterize the hepatic CSCs phenotypes. Susceptibility and resistance to treatments have been widely attributed to cellular heterogeneity and hepatic CSCs in HCC. PD-L1 expression was positively correlated with EpCAM, CD44, and CD133 in breast and colon cancer, but it was negatively correlated with ALDH in cholangiocarcinoma. These four markers are considered CSCs markers in HCC.In vitro,it is beneficial to perform isolation of different (sub)populations from a single HCC cell line, even by single-cell sequencing, to define the preference of the expression of PDL1. Data from one cell line should be compared with others to comprise HCC multiple classifications. The use of animal and variousin vitromodels is also crucial to study the intrinsic PD-L1 in its correlation with hepatic CSCs and cellular heterogeneity. A recently described orthotopic HCC mouse inoculated with PD-L1-expressing liver cells could give more information[215].
In summary, the study on PD-1/PD-L1 immunotherapy could be an emerging and promising approach for HCC therapy. However, the understanding of cancer heterogeneity must be clarified for better selection of patients who are eligible to receive treatment. Comprehensive translational scientific information from cell and animal models, and clinical samples will help the progress of the development and application of immunotherapy in the future.
杂志排行

World Journal of Stem Cells的其它文章
- Epigenetic modulators for brain cancer stem cells: Implications for anticancer treatment
- Mechanisms involved in selecting and maintaining neuroblastoma cancer stem cell populations, and perspectives for therapeutic targeting
- Roles of mitochondrial unfolded protein response in mammalian stem cells
- Stem cell therapies in tendon-bone healing
- Exosomal microRNAs from mesenchymal stem/stromal cells:Biology and applications in neuroprotection
- Bone marrow mononuclear cells for joint therapy: The role of macrophages in inflammation resolution and tissue repair