Mechanisms involved in selecting and maintaining neuroblastoma cancer stem cell populations, and perspectives for therapeutic targeting
2021-07-30AntoniettaRosellaFarinaLuciaAnnamariaCappabiancaVeronicaZelliMichelaSebastianoAndrewReayMackay
Antonietta Rosella Farina, Lucia Annamaria Cappabianca, Veronica Zelli, Michela Sebastiano, Andrew Reay Mackay
Antonietta Rosella Farina, Lucia Annamaria Cappabianca, Veronica Zelli, Michela Sebastiano,Andrew Reay Mackay, Department of Applied Clinical and Biotechnological Sciences,University of L'Aquila, L'Aquila 67100, AQ, Italy
Abstract Pediatric neuroblastomas (NBs) are heterogeneous, aggressive, therapy-resistant embryonal tumours that originate from cells of neural crest (NC) origin and in particular neuroblasts committed to the sympathoadrenal progenitor cell lineage.Therapeutic resistance, post-therapeutic relapse and subsequent metastatic NB progression are driven primarily by cancer stem cell (CSC)-like subpopulations,which through their self-renewing capacity, intermittent and slow cell cycles,drug-resistant and reversibly adaptive plastic phenotypes, represent the most important obstacle to improving therapeutic outcomes in unfavourable NBs. In this review, dedicated to NB CSCs and the prospects for their therapeutic eradication, we initiate with brief descriptions of the unique transient vertebrate embryonic NC structure and salient molecular protagonists involved NC induction, specification, epithelial to mesenchymal transition and migratory behaviour, in order to familiarise the reader with the embryonic cellular and molecular origins and background to NB. We follow this by introducing NB and the potential NC-derived stem/progenitor cell origins of NBs, before providing a comprehensive review of the salient molecules, signalling pathways, mechanisms,tumour microenvironmental and therapeutic conditions involved in promoting,selecting and maintaining NB CSC subpopulations, and that underpin their therapy-resistant, self-renewing metastatic behaviour. Finally, we review potential therapeutic strategies and future prospects for targeting and eradication of these bastions of NB therapeutic resistance, post-therapeutic relapse and metastatic progression.
Key Words: Neural crest; Neuroblastoma; Cancer stem cells; Polyploid giant cancer cells molecular mechanisms; Therapeutic targeting; Tumour microenvironment
INTRODUCTION
Cancer stem cells (CSCs) are subpopulations of aggressive, undifferentiated,multipotent, self-renewing tumour cells with similar behaviour and gene expression patterns to normal stem cells. In pediatric neuroblastomas (NBs), CSCs are considered to be responsible for tumour heterogeneity, genetic instability, therapeutic resistance,post-therapeutic relapse and metastatic progression, making them critical therapeutic targets, the eradication of which will improve therapeutic outcomes. In this review,dedicated to NB CSCs and the prospects for their therapeutic eradication, we provide initial descriptions of the unique transient vertebrate embryonic neural crest (NC)structure and salient molecular protagonists involved in NC formation and migratory behaviour, in order to familiarise the reader with the embryonic cellular and molecular origins and background to NB. We follow this with an introduction to NBs and their potential cellular origins, describe salient molecules, signalling pathways, mechanisms and conditions involved in promoting, selecting and maintaining NB CSC subpopulations, and that underpin their self-renewing, therapy-resistant and metastatic behaviour. We also review potential therapeutic strategies and future prospects for targeting and eradication of these bastions of therapeutic resistance, post-therapeutic relapse and metastatic progression.
THE NEURAL CREST
The unique vertebrate embryonic NC structure was first described by Wilhelm His in 1868 as a band of particular material lying between the presumptive epidermis“Hornblatt” and the neural tube, termed the “Zwischenstrang”, and the origin of spinal ganglia[1]. The NC, composed of multipotent NC cells, appears in humans towards the end of gastrulation at the dorsal edges of the neural folds and neural plate(NP) along the entire neuro-axis[2-5]. Once formed, NC cells undergo epithelial to mesen-chymal transition (EMT), delaminate and migrate throughout the embryo to form pigment cells, myelinating Schwann cells, neurons and glia of the peripheral nervous system, craniofacial skeletal cells including osteoblasts and chondrocytes,mesenchymal cells including connective tissue cells, smooth muscle cells, adipocytes,cardiac cells of the outflow tract, secretory adrenal medulla chromaffin cells, for reviews see[2-6] , and form Schwann cell progenitor cells, a second source of multipotent progenitors, that associate with and migrate along nerve fibres to provide cells for tissue homeostasis and repair during late embryogenesis and in adults[7-9](Figure 1).
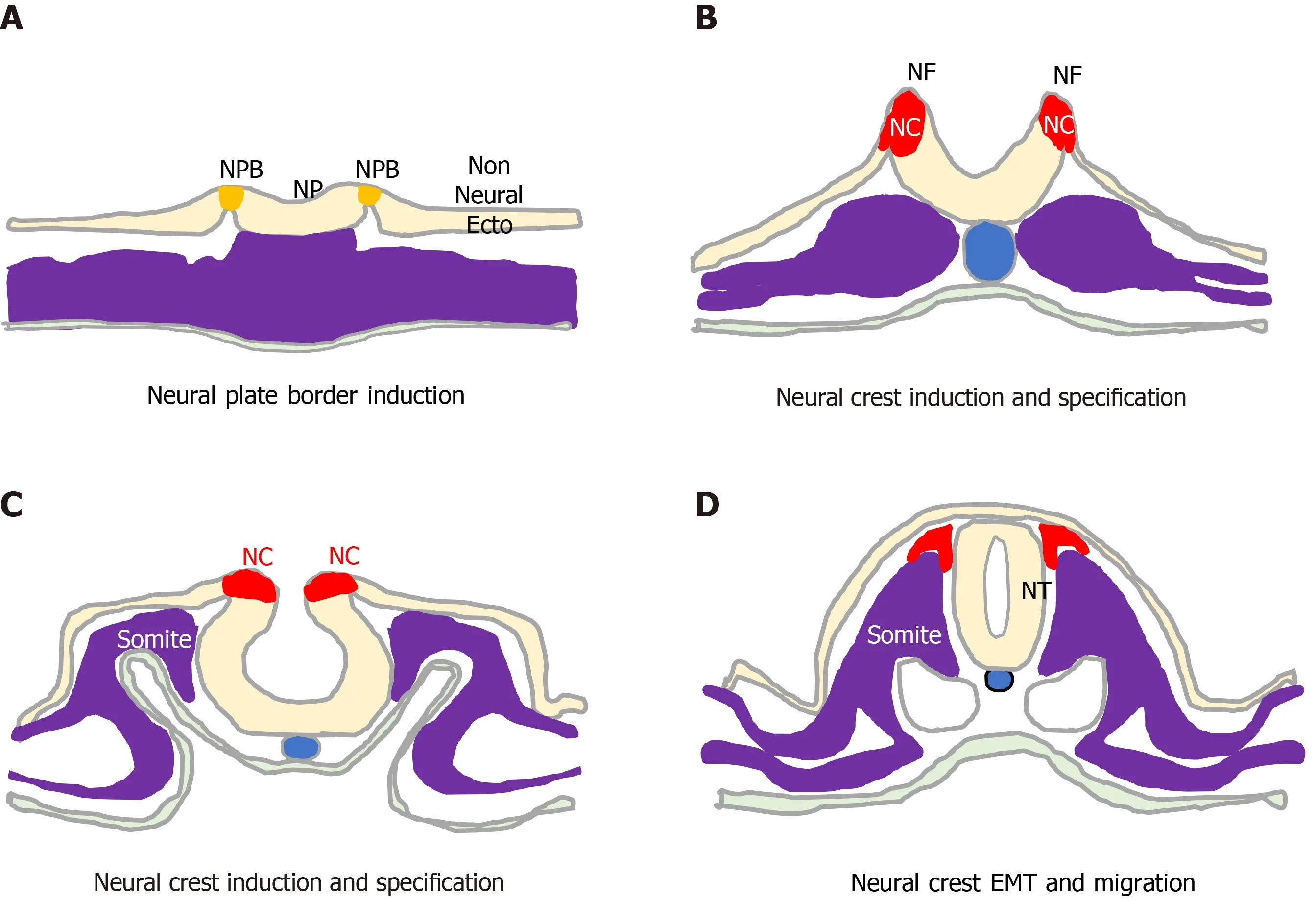
Figure 1 Neural crest formation and behaviour. Initiation, formation and delamination of the neural crest (red). A: This process initiates during early gastrulation with specification of the neural plate border (orange) that separates the neural plate and non-neural ectoderm; B: As the neural folds form, the process proceeds through phases of neural crest (NC) induction; C: NC Specification; and D: Terminates upon neural fold closure and neural tube formation, with NC cell epithelial to mesenchymal transition, delamination and migration. NP: neural plate; NPB: neural plate border; NC: Neural crest; NF: neural fold; NT: neural tube; EMT:Epithelial to mesenchymal transition; non Neural Ecto: non-neural ectoderm.
During development, ectoderm regional patterning forms the NP border and NP,from which the NC, neural tube (NT), nonneural ectoderm and placodes subsequently form[4,10]. This process is regulated by a morphogenic gradient of bone morphogenic protein (BMP) activity within the ectoderm, fibroblast growth factor and wingless/Int-1 (Wnt) signalling pathways[4,10,11]. Subsequent NC specification is an ordered multistep process[4,5], involving Snai2, FoxD3, Ets-1, Sox8, Sox9, Myc, Id2 and later Sox10 transcription factors[12-14], followed by EMT transition[6,10], delamination and NC cell migration throughout the developing embryo, for reviews see[4,6,11,15](Figure 2).
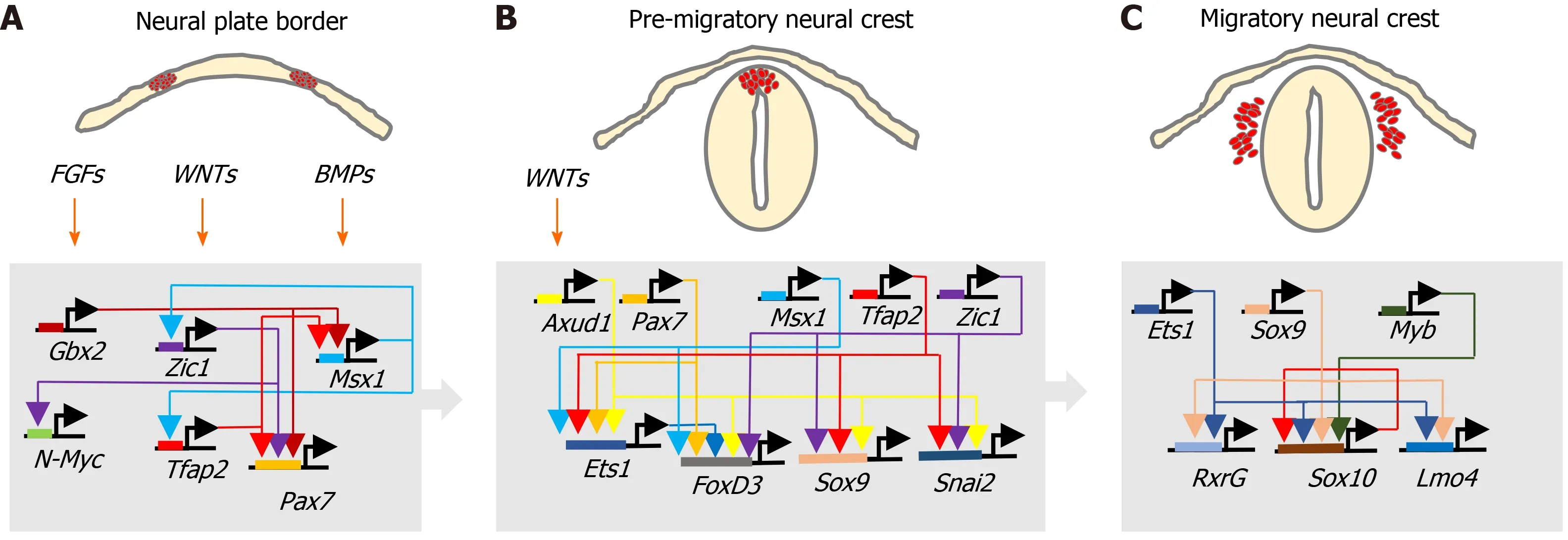
Figure 2 Neural crest transcriptional networks. Interactions between fibroblast growth factor, wingless/Int-1 and bone morphogenic protein signalling pathways and associated transcriptional networks that regulate, A: Neural crest specification within the neural plate border; B: Induction and behaviour of the premigratory neural crest; and C: Migratory neural crest cell behaviour.
NC cell EMT involves the loss of apical/basal polarity, altered expression of type 1(E-cadherin and N-cadherin) and type 2 cadherins (cadherins 6, 7 and 8)[16-19],cytoskeletal reorganisation and proteolytic activity. It is initiated by Snai2, FoxD3,Sox9, Sox10 and Twist transcription factors, regulated by BMP, Wnt-planar cell polarity, Frizzled, RhoA-ROCK signalling in coordination with surrounding tissue morphogenesis[4,6,16], and involves: connexin-43[20], clatherin-mediated endocytosis[21]; cadherin 6B proteolysis[21-23] ; hypoxia-inducible factor (HIF)-a[24-26] ADAMs,γ-secretase and matrix metalloproteinases (MMPs) and matrix components, including collagen, fibronectin, laminin, tenascin, collagen IX, chondroitin-6-sulphate, aggrecan and versecan, which combine to lower intracellular adhesivity and increased motility[27-30]. With respect to the formation of NBs that originate from trunk NC cells, trunk NC cells that delaminate and migrate ventro-laterallyviacontact inhibited locomotion,co-attraction and chemotaxis, accumulate at the dorsal aorta, mix and then form bilateral sympathetic ganglia that go on to innervate various organs and skin[6,28-31].
NC stem cells
The term NC stem cells (NCSCs) was introduced in 1992 by Stemple and Anderson[32], who demonstratedin vitrothat rodent NC cells, in addition to differentiating into autonomic and sensory neurons, glia and smooth muscle cells, also exhibited selfrenewing capacity[32]. At that time, NCSCs were considered a transient populationin vivoand to bein vitroequivalents of embryonic stem cells from blastomeres. NCSCs were subsequently identified in post-natal sciatic nerve, dorsal root ganglion, the gut,bone marrow, cornea, heart, carotid body, dental pulp and periodontal ligament and skin tissues[15,33,34], as a multipotent self-renewing NCSC population resembling embryonic NCSCs in the adult organism[35]. This indicates that, despite the transient nature of the NC, the low self-renewal capacity of NC cells and rapid transition from multipotency to fate and differentiation restriction, undifferentiated NCSCs also populate migrating NC cell streams and post-embryonic tissues, providing an additional population of self-renewing NCSCs that, when necessary, can be called upon to differentiate into specific cell types in response to microenvironmental factors[36,37] and growth factor receptor activation[38], with self-renewal regulated by Wnt and BMP in early migratory NCSCs and later by responses to growth factors[39,40],representing a 4thgerminal layer[15]. Multipotent NCSCs can be isolated from embryos and generated from human embryonic and pluripotent stem cells, with important implications for regenerative medicine and disease modelling[41-43]. Postmigratory NCSCs resemble embryonic counterparts in differentiation capacity, with stemness, migratory behaviour in migrating NC cell populations demonstrated at the single cell level by tracking, and purified cephalic NCSCs have been shown to differentiate into neurons, glia, melanocytes, chondrocytes, osteoblasts and smooth muscle cells[44,45]. A considerable fraction of the NC exhibits an SC phenotype, with fate decisions regulated later by environmental factors, including oxygenation status[46-49], exemplified by: Shh promotion of NC progenitors with mesenchymal skeletogenic, chondrogenic and neurogenic potential; stem cell factor promotion of NCSC survival and melanocyte lineage trophism, when combined with the neurotrophins nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF) and NT3;endothelin-3 promotion of glial and melanocyte progenitor proliferation and survival,and basic fibroblast growth factor promotion of NCSC proliferation[46,47,50,51].
Although there are no specific individual markers for NC cells or NCSCs[4], gene expression patterns that identify potential NCSC populations, include VE-cadherin/CD144, the epidermal growth factor (EGF) family member CFC1/Cripto, transcription factors Pax, Sox10, Hox, mash1, Phox2b; neurotrophic factor receptors p75NTR, RET and EDNRB, and the nerve-related proteins NF, NC-1, E/C8, HNK1, nestin and α4-integrin. RET expression identifies NCSCs within ganglia and is crucial for vagal NC development, P75NTRis used widely to purify NCSCs, Sox10 is considered to be a relatively specific and sensitive NCSC marker, and NSCSsin vitroexpress Sox10,P75NTRand RET[4,15,52,53].
NEUROBLASTOMA
Neuroblastomas (NBs) are small round cell extracranial paediatric tumours that arise during embryonic development from trunk-derived NC cells of the sympathoadrenal lineage and account for approximately 15% of cancer-related childhood deaths. NBs develop anywhere along the sympathetic chain, are more frequent in the abdomen and adrenal medulla, exhibit broad clinical heterogeneity, ranging from spontaneous regression to aggressive metastatic disease and are highly refractory to therapy. Low and intermediate-risk NBs exhibit cure rates of 80%-90%, and < 50% for high-risk disease, with < 10% survival associated with relapsed recurrent disease, for recent reviews see[54,55].
Chromosome aberrations associated with high-risk NB, include hemizygous or homozygous 1p deletions, heterozygous 11q deletions, 17q gains, 5p15.33 rearrangements, and deoxyribonucleic acid (DNA) methylation[56,57]. Although NBs exhibit low somatic mutation rates and no single mutation can explain tumour initiation, activating mutations in the anaplastic lymphoma kinase (ALK) gene characterise familial NBs and 7%-10% of non-familial NBs, activating PTPN11 mutations are associated with approximately 3% of NBs, activating N-Myc mutations with approximately 2% of NBs and activating KRAS mutations with approximately 1% of NBs.Inactivating mutations that are associated with NB include, paired-like homeobox-2 mutations in familial NBs and approximately 4% high-risk NBs, ATRX transcriptional regulator mutations in approximately 2.5% of NBs, p53 mutations in approximately 2% of primary and approximately 10% of relapsed NBs, BRCA2 mutations in approximately 1% of NBs and ARIDIA1A/1B mutations in approximately 3% of NBs[58-63].Despite low level p53 mutation, p53 inactivation is common in NB[64], and several long non-coding ribonucleic acids (lncRNAs) have been implicated in NB progression[65].
N-Myc overexpression combined with activating ALK mutations have been implicated in NB initiation. N-Myc regulates trunk NC cell migration, balances sympathoadrenal progenitor expansion with apoptosis andN-Mycgene amplification characterises approximately 50% of advanced stage high-risk NBs. Mice transgenic for tyrosine hydroxylase-promoted N-Myc expression form NBs in sympathetic ganglia[66], N-Myc overexpression induces NBs in zebra fish[67], and mice transgenic F1174F mutation-activated ALK develop NBs in the presence of high-level N-Myc expression[68]. Tyrosine kinase receptor A (TrkA) and tyrosine kinase B (TrkB) neurotrophin receptors have also been implicated in NB pathogenesis[16,69]. TrkA is required for sympathetic nervous system development and is expressed by NC cells in sympathetic ganglia where it regulates proliferation, survival, differentiation and culling under neurotrophin limiting conditions[16]. TrkA expression in NB associates with favourable prognosis, spontaneous regression and Schwann cell stroma-rich ganglioneuromas[16,69]. However, NBs exhibiting Ip36.2 deletions lose cell surface TrkA expression, in a potential tumour initiating mechanism based upon NC cell escape from neurotrophin-dependent differentiation and apoptosis[70]. Furthermore,oncogenic alternative TrkAIII splicing also represents a potential stress-regulated tumour promoting switch in NB, is exhibited by advanced stage metastatic and relapsed NBs, transforms NIH 3T3 fibroblasts and exhibits oncogenic activity in NB models. Both of which help to explain why TrkA expression is not always associated with a favourable prognosis in NB[16,69,71,72]. Other potential NB susceptibility genes includeLIN28B,CASC15,NBAT-1,BARD1,DDX4,IL31RA,HSD17B12,HACE1,LIN28BandNEFL. Of these,LIN28Binduces N-Myc expression and alters the expression of micro ribonucleic acids (miRNAs), RAN and Aurora A kinase, in a potential NB-inducingLin28B-Ran-Aurora A kinase axis,BARD1polymorphisms promote NB cell proliferation and invasiveness, identifyingBARD1as a potential NB tumour suppressor, and the lncRNAs CASC15 andNBAT1interact with 17q gainassociated Sox9 and USP36 to inhibit NB differentiation[65].
The cellular origins of neuroblastoma:With respect to the cellular origins of NBs, NC progenitors and NCSCs have both been identified as potential origins by their behaviourin vitroandin vivo, by Myc or tamoxifen-inducibleMyc-ERTgene expression patterns in immortalized murine NC progenitor cells engineered to express mutated ALK oncogenes[73,74] and by enforced N-Myc transformation of wild-type NC cells grown from murine NT explants, into NB forming cells[75]. A central nervous system(CNS) neural stem cell-like origin has also been proposed as an origin for some N-Myc amplified NBs, as ectopic N-Myc expression in avian NC cells promotes NC cell conversion to neural stem cells (SCs) that exhibit trunk migration and inadvertent colonization of sympathetic chain ganglia, as a potential primed NC origin for NB,helping to explain gene expression patterns in some N-Myc amplified NBs[76]. A sympathoadrenal progenitor origin for NB is supported by transgenic mouse models of NC targeted N-Myc and ALKF1174Lexpression, which results in perinatal lethality,indicating the NC cannot support oncogenic transformationin vivo, and the absence of the NCSC marker Sox10 and NSCS gene regulatory network expression in human NBs and NB cell lines, contrasting with Sox9 and Sox9-regulated gene expression,consistent with a sympathoadrenal (SA) rather than NC progenitor origin[77].Furthermore, the migratory behaviour of human NB grafts in avian embryonic NC recapitulates the collective migratory behaviour of SA progenitors, avoiding endogenous no go areas and coalescing in sympathetic ganglia and adrenal medulla,which are SA progenitor target sites[78,79]. A potential Schwann cell progenitor origin for some NBs has also been proposed, considering that > 50% of NBs arise in the adrenal medulla and in sympathetic ganglia Schwann cell progenitor target sites, and exhibit distinct features[80]. This suggests different origins for NBs that develop within the sympathetic chain and adrenal medulla, further supported by differences in dissemination and migratory behaviour, illustrated by the migratory behaviour of NB cells transplanted onto the avian embryo NC, which migrate to form primary tumours in sympathetic ganglia, then exhibit secondary migration along nerves and blood vessels, reminiscent of Schwann progenitor migratory behaviour[9,78,79,81]. Malignant Sox10 expressing Schwann cell progenitor subpopulations with stem cell-like features have also been reported in human NBs, connecting adrenergic and mesenchymal compartments through transitions, reminiscent of Schwann cell progenitors[82], and may or may not be involved in the formation of the Schwannian stroma that characterises favourable NBs and benign ganglioneuromas[83,84].Therefore, NBs originate from either NCSCs, SA or Schwann cell progenitors or from NC cells converted into CNS SCs by aberrant N-Myc expression with full transformation within target tissues (Figure 3).
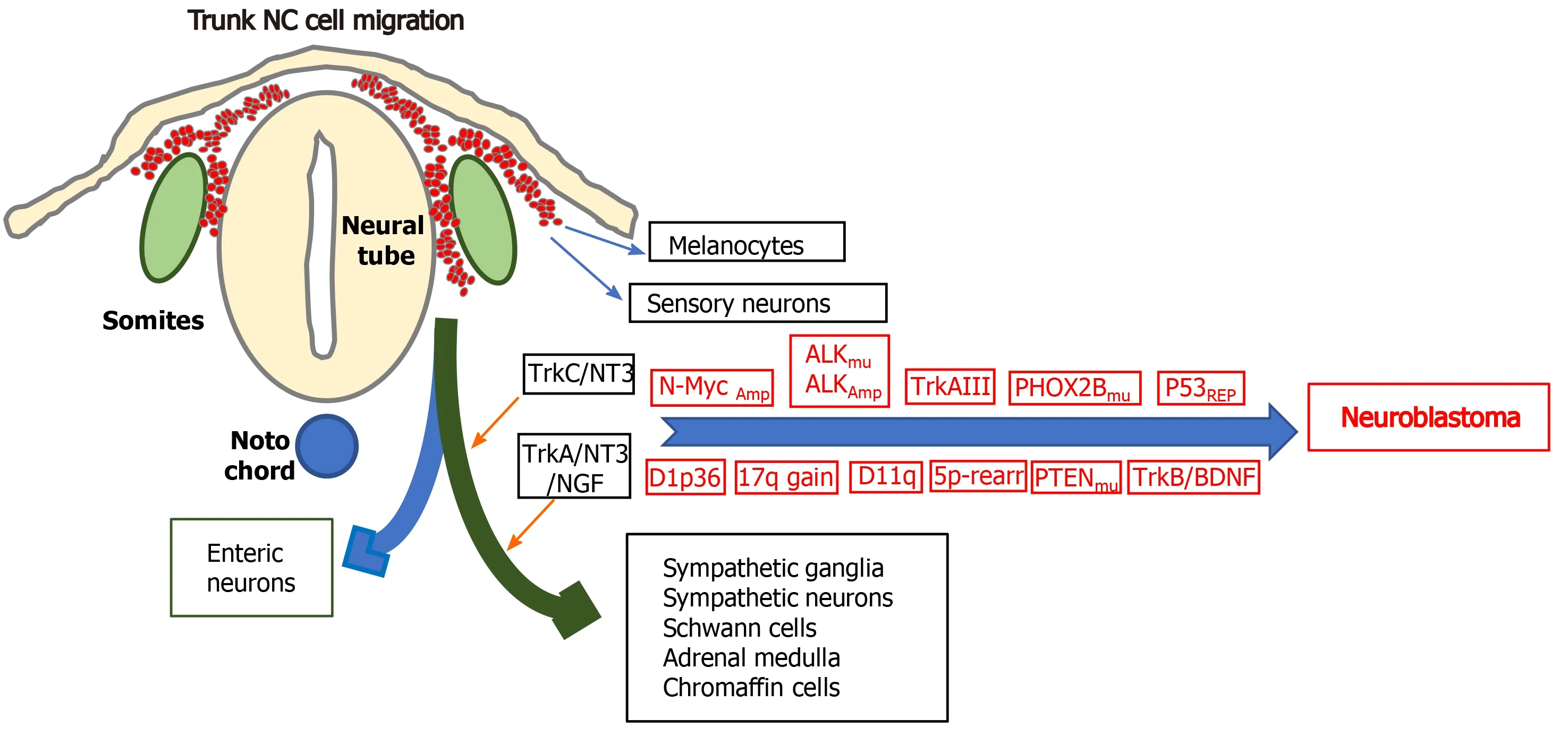
Figure 3 Genetic alterations that characterise neuroblastomas: Illustration of some of the genetic alterations, N-Myc amplification, anaplastic lymphoma kinase amplification and mutation, alternative TrkAIII splicing, p53 functional repression; chromosome 1p36 and 11q deletions; chromosome 17q gains; chromosome 5p rearrangements; mutations in PHOX2B and PTEN and brain derived neurotrophic factor/tyrosine kinase B activation, involved in neuroblastoma formation from trunk neural crest cells of the sympathoadrenal lineage, at sympathetic chain and adrenal medulla target sites, with the differentiated cell types that form from multipotent trunk neural crest cells. NC: Neural crest; ALK: Anaplastic lymphoma kinase.
Neuroblastoma cancer stem cells:Consistent with potential NCSC, CNS SCs,sympatho-adrenal and/or Schwann cell progenitor origins, NC cell multipotency maintenance during migration and reversible phenotypic plasticity at early target sites, NB CSC subpopulations may either represent the original transformed population, or may arise easily through flexible phenotypic plasticity and adaptive stemness signalling within the ever changing tumour microenvironment, as they are only a relative “stone’s throw” away from stemness at the outset[85]. Initial reports of CSC-like subpopulations in NBs, include coordinated expression of stem cell factor and c-kit receptor, which regulate the hematopoietic stem cell niche[86,87] , the characterisation of reversible S (adherent) Schwann cell-like, N (less-adherent) neuronal-like and I (intermediate adherent) neuroblast CSC-like phenotypes in NB cell linesin vitro[88,89], and demonstrate that I-type CSC-like NB cells exhibit higher malignant clonogenic and tumourigenic activityin vitroandin vivo,reminiscent of malignant NCSCs, and in NBs correlate with disease progression[89-92]. I-type multipotency has been demonstrated by reversible spontaneous or induced differentiation to N-type and/or S-type phenotypes[88,93], and an I-type sympathoadrenal CSC-state suggested by CD133, c-kit (CD117), Sox2, GPCRC5C, NOTCH-1, p75NTR, TrkA, TrkB and tyrosine kinase C (TrkC) expression[92,94]. I-type CSC-like NB cells also exhibit enhanced levels of activated guanosine triphosphate (GTP)-bound N-Ras, compared to S or N phenotypes, associated with reduced expression of the Ras GTPase activating factor neurofibromin, resulting in augmented tumourigenic activity[95]. More recently, a PHF20/PARP1 axis has been identified in NB I-type cells that modulatesH3K4trimethylation, inducing Sox2 and Oct4 expression, leading to Nanog expression, Wnt signalling and N-Myc expression, involved malignancy regulating CSC self-renewal and EMT traits[96]. CSC-like SP NB populations, detected by double-labelling of neuronal specific neurofilament and calcium binding protein S100A6 have also been identified, that exhibit Hoechst dye exclusion, express GD2, cKit, CD133, CD71, CD56 and ABCG2, comprise from 0.8% to 50% of total viable cell numbers in NBs, and are present in high-risk unfavourable primary NBs and bone marrow metastases[97-99]. In micro-array analyses, I-type CSC-like NB cells exhibit constitutive elevated expression of CD133, c-kit, GPRC5C, Notch1, Pigf2, TrkA, TrkB, TrkC and p75NTR[89,92]. CD133 and c-kit are considered to be markers of NB CSCs, CSC survival, growth regulation and poor clinical outcome[12,100]; GPRC5C is considered to indicate neuroectodermal origin and a differentiated embryonic stem cell state; Notch is involved in neural SC maintenance[101-103]; Pigf2 is involved in tumour angiogenesis growth and survival[104], and neurotrophin receptors are involved in NB growth, invasion and survival[16,100]. Other reverse transcription-polymerase chain reaction studies have reported increased CD133, Sox2, Bmi1 and Msi1 expression in immature NB CSC-like cells[105], and genome-wide microarray analysis of CSCs enriched from NB cell lines have identified Wnt, Notch, Shh and TGFβ signalling pathways in self-renewing CSC-like phenotypes, with TGFβ activation associated with repression of SMAD6 and SMAD7 TGFβ signalling repressors, Shh signalling associated with expression of Patched, Gli1 and smoothened, and CD133 and CD15 expression enriched in CSC-like neurospheres[106].
MOLECULES AND MECHANISMS THAT PROMOTE, SELECT AND MAINTAIN NB CSCS
CSC markers differ between tumour types, many are also expressed by normal cells and cell surface molecular markers can be used for targeted therapy. It is important,therefore, to understand markers and patterns of marker expression that specifically define NB CSC populations, since private highly specific functional CSCs cell surface markers represent the best targets for monoclonal antibody and chimeric antigen receptor T-cell immunotherapy (CAR-T) therapy, and CSC-specific intracellular markers and signalling pathways may provide targets for small molecular inhibitors.
Cell surface and membrane-associated molecular protagonists
CD133, CD44 and CD24: The 5 transmembrane domain cell surface 120 kDa glycoprotein CD133 (AC133/Prominim-1) was originally identified as a haematopoietic stem cell marker[107] and later detected in epithelial progenitors and implicated in brain tumour CSC self-renewal and tumour initiating capacity[108].CD133 is considered to be a NB CSCs marker, is detected in approximately 0.1% of NB cells in NB tissues, and CD133+NB cells exhibit self-renewal capacity, multipotency and enhanced chemoresistance, and predict disease outcome in NBs[109,110]. I-type CSC-like NB cells exhibit high-level CD133 expression[91], which regulates proliferation, tumourigenic activity and represses differentiation induced by inhibiting RET signalling[100].
The transmembrane glycoprotein CD44 mediates cell-cell interactions and cellular attachment to extracellular matrices by binging hyaluronic acid. CD44 is expressed as a variety of isoforms and in NB is a strong prognostic marker, together with N-Myc.Both CD44 expression and lack of expression have been associated with tumour initiating NB CSC-like properties[111,112]. CD44 negative NB populations express higher levels of the tumour initiation marker CD24, and exhibit a gene expression pattern characterised by reduced p21Cip1, p16INK4a and p15INK4b expression,consistent with uncontrolled cell cycle progression, reduced expression of apoptosis factor NAIP, Bcl-xl, Bax and IRF1, consistent with apoptosis evasion and are tumourigenic in NOD/SCID mice, in which CD44 expression is inversely related to metastatic propensity[111]. In contrast, high level CD44 expression is associated with low survival in high-grade NB and NB cell populations enriched for CD44 expression behave like multipotent undifferentiated self-renewing NC-like progenitors, are more invasivein vitro,and more tumourigenic and metastaticin vivo. Furthermore, CD44+/Nestin+/S100b-NB cells have been reported to be more aggressive, tumourigenic,undifferentiated and more NC-like compared to CD44+/Nestin-/S100b+Schwan-like cells[112].
The GPI-anchored CD24 cell surface sialo-glycoprotein is expressed by differentiating neuroblasts, is crucial for neural development and is involved in neurite outgrowth and neurogenesis. CD24 interacts with β1-integrins, activates mitogenactivated protein kinase (MAPK) through focal adhesion kinase and Src kinases, is implicated in NF-κB, Notch and Shh signalling[113] and promotes metastasis by interacting with P-selectin on activated platelets and endothelial cells[114,115]. CD24+/CD34+enriched NB populations are more tumourigenic in mice, CD44-CSC-like tumour initiating NB cells express high levels of CD24[111] and CD24+CSC-like tumour initiating cells are present in bone marrow NB metastases, suggesting that CD24 enriches the tumourigenic potential of CSC-like tumour initiating NB cells within the bone marrow[99].
Delta-like 1 homolog and prohibitins:The development-regulated transmembrane protein delta-like 1 homolog (DLK1), which regulates stem/progenitor cells, is overexpressed in NBs and is robustly expressed in undifferentiated NB cells[116,117]. In NB cells small interfering ribonucleic acid (siRNA) inhibition of DLK1 expression promotes spontaneous neuronal differentiation, decreases clonogenic and colony forming capacity and suppresses tumourigenicity, and vice versa for DLK1 overexpression[116,117]. HIFs bind and activate the DLK1 promoter[117], linking DLK1 expression to tumour hypoxia and oncogenic HIF-1 and HIF-2 activation, explaining the localisation of DLK1+NB cells to hypoxic niches[116]. DLK1 also interacts specifically with prohibitins (PHB) 1 and 2, regulating mitochondrial function and reactive oxygen species (ROS) productionviaa DLK1/PHB axis involved in NB CSC self-renewal and clonogenic capacity[118]. Conversely, high DLK1 expression is associated with NB cells arrested in relatively late-stage chromaffin lineage differentiation[119].
L1-CAM:The neural cell adhesion molecule L1-CAM regulates neuronal growth,survival, migration, axonal outgrowth and neurite extension[120], and has been implicated in CSC marker upregulation and aggressiveness in gliomas. Although L1-CAM expression has been reported to associate with better prognosis in NB[121], L1-CAM knockdown in NB cells down-regulates N-Myc expression, up-regulatesPTENexpression, and inhibits self-renewal, tumour sphere formation and migration,suggesting that L1-CAM may indeed play an important role in NB CSC-like behaviour[122].
ABC transporters:Members of the membrane-associated adenosine triphosphatebinding cassette ABC transporter super-family promote molecular transport across extracellular and intracellular membranes, and are important drug-efflux pumps that increase multidrug-resistance[123,124]. ABCG2 and ABCA3 are co-expressed in Hoechst dye excluding CD133+SP CSC-like NB cells that exhibit enhanced chemoresistance and tumourigenic activityin vivo[97,124-126].
TRPM7:The transient receptor potential cation channel family member and kinase TRPM7 regulates mechanical cellular interactions within microenvironments by interacting with the cellular cytoskeleton, integrating adhesion and differentiation responses with microenvironmental stimuli. TRPM7 expression is essential for maintaining NC cell multipotency, contributes to NB by disrupting NB cell maturation and preserving progenitor CSC-like features, and is promoted by N-Myc in NB cells[127,128]. TRPM7 expression is closely associated with NB CSC-like cell migratory and metastatic behaviour[129], controls developmental transcription through SNAI2,helping to maintain progenitor-like features[130,131], enhances therapeutic resistance[132] and may also promote metastasisviaHsp90a/uPA/MMP-2 signalling[133].
Lamin A/C:The nuclear membrane-associated protein lamin A/C regulates nuclear stability. Knockdown of lamin A/C expression in NB cells promotes aggressive, selfrenewing tumour-initiating CSC-like populations that exhibit a more drug-resistant phenotype[134], increases CSC marker Oct4, Nanog, Sox2 and N-Myc, CD133, Sox2 and SSEA4 expression, and enhances neuro sphere forming capacity[135].
Growth factors, receptors and signalling pathways
Cryptic family member CFC1:The epidermal growth factor family member CFC1 is also considered a potential NB CSC marker, involved in NB cell growth, colony and tumour sphere formationin vitroand NB xenograft tumour formationin vivo. CFC1 inhibits NB differentiation induced by activin A and smad-2 phosphorylation,implicating collaborative signalling between EGF and CFC1 in NB[136].
Frizzled/Wnt signalling and LGR5/GRP49:Wnt signalling is critical for NC induction, specification, EMT and migration, occursviacanonical and non-canonical Wnt pathways, and has been closely implicated in NB progression[137,138]. In the canonical pathway, Wnt ligands bind Frizzled receptors inducing nuclear translocation of β-catenin, which binds and activates Tcf/Lef transcription factors, whilst blocking Tcf/Lef target gene repression, whereas non-canonical pathways include planar cell polarity and calcium mobilisationviaG-protein activation. In NBs, Frizzled Wnt receptor Fzd6 expression predicts poor survival and marks rare HIF1/2a-positive CSC-like tumour initiating NB cell populations located in hypoxic tumour areas[139].Fzd6 positive NB cells share biological features with CSCs including neurosphere formation, increased invasive capacity and resistance to chemotherapeutic agents,Fzd6 is also required for non-canonical Wnt pathway gene expression and is implicated in aggressive NB behaviour, consistent with a role in promoting and maintaining NB CSC-like subpopulations[140]. Wnt pathway promotion of NB CSCs also involves SLC34A2 transcription factor, which negatively correlates with overall and relapse-free survival in NB patients, promotes NB CSCs by binding the miR25 promoter and activating miR25 expression, which binds the Gsk3β messenger RNA(mRNA) 3’-UTR, reducing Gsk3β protein expression, resulting in Wnt signalling[141].The genotoxic agent doxorubicin also enhances Wnt signalling in NB cells in association with up-regulated Frzd4 and 6 expression, implicating the DNA damageresponse in NB CSC-like states[140] and subsequent chemoresistance[142,143].Hyperactivation of the Wnt/β-catenin pathway in NB cells is also promoted by NEU4 Long sialidase, which enhances proliferation, G1 to S phase transition and promotes a more undifferentiated CSC-like phenotype, potentiallyviaMyc, Nanog, Oct4, CD133 and Nestin pluripotency gene expression, identifying NEU4L as a novel potential therapeutic NB CSC target[144]. The SC marker LGR5/GPR49 also promotes canonical Wnt signalling and is overexpressed in advanced stage N-Myc amplified and ALKmutated NBs. LGR5/GPR49 knockdown induces NB cell apoptosis in association with diminished MAPK signalling, identifying LGR5/GPR49 as a novel potential NB CSCs therapeutic target[145,146].
Neurotrophin receptors P75NTR, TRKA, TRKAIII, TRKB, TRKC and sortilin:Neurotrophins NGF, BDNF, NT3 and NT4/5 and their receptors are critical for the development and maintenance of vertebrate central and peripheral nervous systems,and have been implicated in NB regression and metastatic progression. NT receptors include: the tropomyosin-related tyrosine kinases TrkA that binds NGF and NT3; TrkB that binds BDNF and NT4/5; TrkC that exclusively binds NT3; CD271/p75NTRthat binds mature NGF with low affinity and all pro-form NTs with high affinity, and sortilin that binds mature NGF and pro-form NGF, BDNF and NT-3. NT/TrkA activation inhibits default apoptotic programmes to promote NT-dependent survival through PI3K/Akt/NF-κB signalling and Bcl-2 family induction and in the absence of NTs, TrkA and TrkC, but not TrkB, induce apoptosis through a CD271/p75NTRdependent mechanism, identifying TrkA and TrkC as dependence receptors. TrkC is the only NT receptor expressed during early embryogenesis and during neurulation is detected in the NT and NP anlage, and in both pre-migratory and migrating NC cell subsets. NT3/TrkC activation is required for NC cell survival at target sites and,during NC migration, somites express NT-3 and sympathetic neuroblasts and neurons express NT3 and TrkC, promoting neuroblast survival, proliferation and subsequent differentiation. This temporary effect declines and switches to NGF-dependency,associated with reduced TrkC expression and increased TrkA and CD271/p75NTRexpression, regulated in part by autocrine NT3 activity. NT3/TrkA interactions continue to promote sympathetic neuroblasts, neuron survival and target organ innervation, then changes gradually to dependence upon NGF/TrkA interaction[16,69].
CD271/p75NTRis a NC SC marker, and self-renewing CD271/p75NTR+neural SCs coexpress TrkA, TrkAIII, TrkB and TrkC. In general, NB expression of CD271/p75NTRis associated with favourable outcome and CD271/p75NTRexhibits tumour suppressor function in NB models, inducing differentiation, promoting apoptosis and reducing tumourigenic activity. NB CSC-like cells, however, express CD271/p75NTR, CD133+/CD271/p75NTR+self-renewing, clonogenic NB cells produce more malignant tumours,and CD271/p75NTRhas been closely linked to migratory and invasive behaviour of CSCs, implicating CD271/p75NTRin promoting malignant NB CSC-like behaviour[147].
Studies on NT receptors in NB initiated with the detection of an inverse relationship between high N-Myc and low TrkA expression is associated with poor prognosis and mid to high TrkA expression in non-N-Myc amplified disease[148]. Low TrkA expression combined with N-Myc amplification and expression, in general, characterises unfavourable NB but N-Myc amplified NBs also exhibit heterogeneity in TrkA expression, with a small number also exhibiting high TrkA expression, indicating that a more complicated relationship between N-Myc and TrkA in NB[16,69]. In general,moderate to high TrkA expression distinguishes non N-Myc and N-Myc amplified NBs but does not distinguish prognostic groups in non-N-Myc amplified NBs. Low TrkA expression in NBs may relate to a non-TrkA expressing sympathoadrenal cell origin in coalescing sympathetic ganglia, paraganglia or adrenal medulla anlage during development or N-Myc repression of TrkA expression post-transformation through promoter methylation. Conversely, high TrkA expression in NB may relate to an undifferentiated TrkA expressing NC progenitor origin within the sympathetic chain or adrenal primordia or to induction post-transformation by either NTs, growth factors and/or cytokines. In NB models, fully spliced TrkA receptors exhibit tumour suppressor activity and restore NGF-induced neuronal NB cell differentiation,viaIGF2, c-Src, PKCe and Ras/MAPK/Erk signalling, reduce angiogenic factor expression, angiogenesis and tumourigenicity and promote genetic stability. TrkA also induces p53-dependent NB cell apoptosis through CCM2, Erk and caspase-7. NB cell responses to NT/TrkA activation are optimized by CD271/p75NTR, and TrkA/CD271/p75NTRco-expression in NB carries a good prognosis[16,69,148]. The discovery of the oncogenic alternative TrkAIII splice variant in advanced stage NBs and NB cell lines, however, challenges the hypothesis of an exclusively tumour suppressing role for TrkA in NB. TrkAIII alternative splicing is development and stress-regulated and generates an oncogenic TrkAIII isoform that transforms NIH3T3 cells and exhibits oncogenic activity in NB models[69,71,72]. In NB cells, TrkAIII expression induces centrosome amplification and increases genetic instability[149], promotes stressinduced aerobic glycolysis[150], enhances angiogenic factor expression, impedes NGF/TrkA signalling through Ras/MAPK, induces a survival adapted endoplasmic reticulum (ER)-stress responseviaretrograde ERGIC to ER recyclingin vitro[72,151],and enhances tumour-associated angiogenesis, tumourigenesis and metastatic capacityin vivobut does not restore NGF responsiveness nor induce NB cell differentiation or apoptosis[69,71,72].A potential role for TrkAIII in NB CSCs comes from observations that TrkAIII increases mitochondrial Superoxide dismutase 2 (SOD2) expression and activity in NB cells, enhancing resistance to ROS-induced death within the context of CSC-like phenotype[152]. Furthermore, TrkAIII promotes centrosome amplification,and microtubule polymerization resulting in polyploid and aneuploid de-differentiated anaplastic phenotypes[153], implicating TrkAIII in selecting and maintaining CSC-like NB subpopulations within stressful pathological and therapeutic tumour microenvironments[72].
TrkB has also been implicated in NB pathogenesis, progression and therapeutic resistance[16,154-156], exhibits a positive correlation with N-Myc amplification and expression, and is stimulated by activated Erb-B in NB cells. Unfavourable NBs express BDNF providing a potential autocrine/paracrine survival mechanism and TrkB is transiently expressed by sympathoblast subpopulations during sympathetic nervous system development, providing a potential origin for TrkB expressing NBs,with BDNF expression acquired at a later stage[157]. TrkB expression is increased by HIF-1 suggesting that tumour hypoxia may promote TrkB expression in NBs, and BDNF/TrkB interaction increases NB cell survival, chemoresistance, invasive capacity,angiogenesis factor expression, angiogenesis and metastatic behaviour[16,156,157].Recently, an engineered TrkB receptor, devoid of Ig-like domains, analogous to TrkAIII, has been shown to be oncogenic[158], although a similar endogenous isoform(s) has not yet been described in NB.
TrkC expression in NB is associated with a favourable outcome. However, high level NT-3 and TrkC co-expression has been identified in a subset of advanced stage IV NBs, providing a potential paracrine/autocrine survival and proliferation mechanism for selection in tissues that do not express NT3, similar to migrating NCderived sympathoblasts, as a potential cellular origin for this NB subset[16]. In further support of NT receptor involvement in NB stemness, I-Type CSC-like NB cells exhibit high-level TrkA, TrkB, TrkC and CD271/p75NTRexpression[92].
ROR1:The receptor tyrosine kinase-like orphan receptor ROR-1 is related to CSC-like phenotypes[159], is expressed across all NB stages, correlates with survival and prognosis, and targeted anti-ROR-1 therapy results in additive cytotoxicity to NB cells,identifying ROR1 as a novel regulator of the NB CSC compartment and potential therapeutic target[160].
Apoptosis signal regulating kinase 1:The apoptosis signal-regulating kinase apoptosis signal regulating kinase 1 (ASK1) is a member of the MAPK family and activates JNK and p38MAPK in response to ER-stress, oxidative stress and calcium influx. ASK1 is co-expressed and associates with TLX/NR2E1 in CSC-like NB SP cells,particularly in hypoxic conditions within the CSC niche. ASK1 phosphorylation stabilises TLX/NR2E1, resulting in HIF-1α induction and VEGF-A expression,contributing to angiogenesis and NB CSC survival[161].
Transcription factors and regulators
STAT3 granulocyte colony-stimulating factor receptor and CD114:STAT3 transcription factor is critical for NC specification and promotes an undifferentiated NC phenotype. Loss of STAT3 expression results in apoptosis and NC marker Sox10 and Snai2 repression[162]. STAT3 maintains CSC populations in bladder, colon, and hepatocellular cancers and malignant gliomas[163-166]. STAT3 activity is induced by granulocyte colony-stimulating factor (G-CSF) receptor activation and G-CSF receptors are expressed by neuronal and NC-derived cell types, in which STAT3 promotes SC survival and expansion. Highly tumourigenic multipotent CD114+NB cells, that exhibit self-renewal, also exhibit STAT3 activation in response to G-CSF. G-CSFsensitive cells in primary and relapsed NBs are considered to represent a premigratory NC cell population and express multiple miRNAs with validated targets in p53, reprogramming[167-169], SC cell-cycle regulation and neuronal differentiation pathways[170,171], providing a paracrine G-CSF-mediated mechanism for expanding highly tumourigenic CD114+CSC-like NB subpopulations, which is of particular relevance to the therapeutic use of G-SCF in NB [172,173].
FOXD3 and FOXM1:PDFG, Wnt and BMPs signalling regulate NC induction and specification through transcription factors, including FoxD3 (Tenget al[174], 2008). The forkhead family transcriptional repressor FoxD3, originally characterised in embryonic SCs and multipotent NC cells[175,176], is considered to be an early NC lineage marker that specifies fate by upregulating HNK1 and Slug expression, involved in maintaining the undifferentiated state of migrating NC cells and self-renewal potential[174]. In NB, FoxD3 acts as a tumour suppressor, regulating the expression of N-Myc downstream regulated 1 and downstream genes. FoxD3 repression promotes NB tumourigenic, invasive, metastatic and angiogenic activityin vitroandin vivo, and is associated with NB CSC phenotypes[177]. In contrast, FoxM1 is a proliferation-specific transcription factor, the knockdown of which induces NB cell differentiation,associated with Sox2 and Bmi1 repression. FoxM1 directly activates Sox2 expression in NB cells and regulates both Sox2 and Bmi1 expression, required for neural stem cell self-renewal and maintenance of an undifferentiated NB CSC-like state[176,178].
Bmi1 and Kif1Bβ:The polycomb group transcriptional repressor Bmi1, originally identified in murine lymphomas as an oncogenic partner of c-Myc, forms part of Polycomb repressor complex 1, involved in gene repression through chromatin modification[179,180]. Bmi1 is required for NCSC self-renewal, CSC self-renewal and tumourigenicity[181,182] and Bmil knockout reduces NCSC self-renewal and results in progressive loss of NCSCs in mice[183]. NB CSCs express Bmi1, and the Bmi1 promoter binds and is activated by E2F-1 transcription factor[184]. Knockdown of Bmi1 expression in NB cells induces differentiation and growth suppression, and the role of Bmi1 in NB tumourigenesis has been linked to FoxM1, which activates the expression of both Sox2 and Bmi1[178]. In I-type CSC-like NB cells, Bmi1 is required for self-renewal but also influences lineage commitment. Bmi1 repression promotes Stype differentiation, high Bmi1 expression induces N-type differentiation and Bmi1 over-expression induces neuronal differentiation, implicating fine concentrationdependent Bmi1 control of the delicate balance between NB CSC self-renewal and differentiation[182]. Bmi1 transiently inhibits the p53 response to N-Myc-induced oncogenic-stress in perinatal NB precursor cells by directly binding and promoting p53 ubiquitination and degradation, in a general mechanism for p53 inactivation in NBSC-like progenitors, susceptible to N-Myc oncogenesis during embryonal development[185]. Bmi1 is also a N-Myc target-gene involved in repressing expression of the NB tumour suppressors TSCL1 and Kif1Bβ, with implications for NB pathogenesis[186,187], and promotes IkBα degradationviaubiquitination within the context of the SCF complex, inducing CSC survival NF-κB signalling, tumourassociated inflammation, proliferation, MMP-9 expression and EMT[188-191].
The kinesin Kif1Bβ acts downstream of EglN3 to induce neuronal apoptosis and exhibits loss of function mutation or loss associated with Ip36.2 deletion in NBs,resulting in apoptosis evasion[192]. Kif1Bβ is also required for neuroblast differentiation and in mouse sympathetic neuroblasts deleted of Kif1Bβ and human NBs that lack Kif1Bβ, neural differentiation is ablated. Loss of Kif1Bβ expression contributes to undifferentiated aggressive NBs, characterised by high-level Sox10 expression, loss of cell surface TrkA expression, and up-regulated expression of ALK ligands and suspected MDK and NHLH2 NB oncogenes[70]. Furthermore, NB cells that lack cell surface TrkA, due to Kif1Bβ deletion or mutation, do not exhibit NGF-mediated differentiation and in the absence of neurotrophins, escape TrkA-induced apoptosis,compounding the Sox10 expressing de-differentiated NB CSC-like state[70].Bmi1/Kif1Bβ involvement in NB CSC induction and maintenance has also been associated with a novel alternative extra-short form NF-YAx splice variant subunit of the ubiquitous NF-Y transcription factor. NF-YAx was originally identified in primary human NBs, is expressed during murine embryonic development at times corresponding to sympathetic neuroblast culling and is induced in NB cells by the genotoxic chemotherapeutic agent doxorubicin. NF-YAx expression in NB cells represses Bmi1 expression and enhances Kif1Bβ expression, resulting in necroptosis, suggesting a NB tumour suppressor function. However, chronic NF-YAx expression also selects tumourigenic NB cells that are resistant to NF-YAx-induced cytotoxicity, do not exhibit NF-YAx-induced Bmi1 repression and Kif1Bβ induction, and exhibit a CSC-like expression signature characterised by enhanced CD117, p75NTR, Nanog, Nestin, Sox2 and EglNL3 mRNA expression, indicating that NF-YAx selects NB CSC phenotypes by eliminating cells sensitive to NF-YAx-induced necroptosis, in a potential DNAdamage-induced CSC selection mechanism, with implications for NB genotoxic chemotherapy[193].
N-Myc, cMyc and ALK:Myc/Max complexes inhibit ESC and iPSC differentiation and have been implicated in the epigenetic reprogramming that enhances CSC phenotypes[194]. Both N-Myc and mutated ALK are important NB oncogenes and collaborate to promote NB progression. In embryonic sympathetic neuroblasts, N-Myc overexpression promotes proliferation but does not sustain survival, and long-term ALKF1174Lactivation promotes cell cycle arrest, differentiation and survival of postmitotic neurons, in association with NEFM, RET and VACHT expression and decreased BMF, BIM, TH, CDC25A and CDK1 expression. N-Myc and ALKF1174Lcoexpression maintain neuroblast proliferation and promote a different differentiated phenotype, characterised by SKP2, CCNA2, E2F8 and DKC1 expression. This is consistent with initial steps towards NB development, and is supported by demonstration that N-Myc and ALKF1174Lare sufficient to drive NB development from NC progenitor cells[73,195,196]. N-Myc also forms a positive feedback loop with its cis-antisense gene N-Cym, co-amplified with N-Myc, and Oct4 transcription factor[197]. This contributes to a more CSC-like aggressive phenotype in N-Myc amplified NBs, as N-Cym stabilises N-Myc, resulting in Oct4 and CSC-related Nanog, Sox2 and Lin28 expression[198]. N-Myc also promotes survival by up-regulating components of the error-prone non-canonical alternative nonhomologous end-joining DNA repair pathway, resulting in increased DNA repair activity to overcome DNA damageinduced death. This behaviour is shared with NCSCs, consistent with a NCSC status of N-Myc amplified NB cells[199]. N-Myc also promotes lysine 4 histone H3 trimethylation in stem cell-related genes, and in microarray studies, N-Myc amplified NBs exhibit Lif, Klf2, Klf4 and Lin28b ESC-related factor expression, with N-Myc observed to directly bind to the Lif promoter, implicating N-Myc in the regulation of overlapping SC-related gene expression, CSC-like pluripotency and self-renewal in NB[200]. Glutamine is also involved in N-Myc regulation of NB CSCs, as glutamine deprivation leads to N-Myc/c-Myc cooperation, resulting in CD133 expression in NMyc amplified NB cells, associated with changes in acetaldehyde dehydrogenase(ALDH) activity[201].
NF-Y:The ubiquitous heterotrimeric transcription factor NF-Y binds inverted CCAATboxes in approximately 70% of gene promoters and is composed of NF-YA, NF-YB and NF-YC subunits[202-204]. Alternative splicing of the NF-YA subunit has been implicated in cellular staminality, differentiation, apoptosis and transformation, and the short alternatively spliced NF-YAsisoform forms part of the SC transcriptional circuitry, predominates in ESCs and is lost upon differentiation, whereas fully-spliced long-form NF-YAl promotes differentiation and NF-YA loss induces senescence or apoptosis[205-207]. Alternative NF-YAssplicing is promoted by oncogenes and converts tumour suppressing, differentiation promoting NF-Y complexes predominated by NF-YAl into tumour and CSC promoting complexes predominated by NFYAs[208,209]. Recently, a novel developmental and doxorubicin-regulated extra shortform alternative NF-YAx splice variant has been identified in primary stage 2 and stage 3 NBs[210,211]. This novel isoform forms functional NF-Y complexes but has lost capacity to bind the ubiquitous cancer-associated transcription factor SP1[211], characterising NF-YAx as a functional NF-Y modifier. NF-YAx expression in NB cells promotes Kif1Bβ-dependent necroptosis and is expressed during murine embryo development at times corresponding to Kif1Bβ-dependent sympathetic neuroblast culling, consistent with a potential tumour suppressor function. However, NF-YAx also selects CSC-like subpopulations resistant to NF-YAx-induced death that exhibit enhanced resistance to the genotoxic agent doxorubicin, which also promotes alternative NF-YAx splicing, implicating NF-YAx as a potential protagonist in genotoxic drug-induced selection of NB CSC phenotypes and subsequent post-therapeutic relapse[210].
Paired related homeobox 1 and notch-3:Cell types identified in NBs include interconverting de-differentiated, mesenchymal, multipotent NC-derived progenitor-like,adrenergic and intermediate cell types that exhibit distinct super enhancer and associated transcription factor networks and divergent gene expression profiles[212,213]. Expression of the embryonal-restricted transcription factor paired related homeobox 1 (PRRX1) in mesenchymal tissues, reprogrammes adrenergic NB cell super enhancer and mRNA landscapes towards the NC-derived progenitor-like mesenchymal cell type, enhancing chemoresistance, implicating PRRX1 in promoting CSClike states and helping to explain the enrichment of PRRX1+cells in post-therapy recurrent NBs[212]. NOTCH-3 also induces adrenergic transcriptional reprogramming to a multipotent, NC-derived progenitor, mesenchymal de-differentiated stateviagenome-wide remodelling of the H3K27ac landscape, in a NOTCH feed-forward loop,that also maintains the mesenchymal NB CSC-like state[214]. Furthermore, hypoxia induces Notch signalling in NB cells with implications for NB cell de-differentiation[215].
Repressor element 1 silencing transcription factor, MUSASHI1 and C-KIT/CD117:Repressor element 1 silencing transcription factor (REST) zinc finger transcription factor is a master repressor of neuronal programmes, helps to maintain de-differentiated SC and CSC states and promotes genomic instability, contributing to cellular transformation[216,217]. In NBs, REST exhibits oncogenic activity and high REST expression is associated with poor prognosis, supporting a potential role in promoting NB CSC-like behaviour[218-220].
The mRNA-binding protein Musashi1 is a critical regulator of SC self-renewal and has been implicated in CSC-like self-renewal capacity in NB by promoting target mRNA translation and stem cell-fate transition[105,221]. Although the signalling mechanisms involved in Musashi1 function in stem/progenitor cells are unclear,Musashi1 is regulated by phosphorylation and by MAPK signalling in oocytes, and exhibits functional duality by promoting translation and repressing mRNA targets, in a similar way to Musashi2, which represses TGFβR1 mRNA, whilst activating translation of SMAD3 mRNA in proliferating K562 myelogenous leukemia cells[222].
The c-kit/CD117 stem cell factor receptor tyrosine kinase is a SC and CSC marker[91,223], and has been reported to be one of seven genes exhibiting elevated expression exclusively in NB CSCs[92].
JARID1B:The H3K4me3 histone demethylase JARID1B is involved in ESC fate and plays important roles in developmental gene expression and neural differentiation. In NB, JARID1B expression enhances CSC-like behaviour, therapeutic resistance and enables transit between distinct CSC-like cell populationsviathe PI3K pathway[224].JARID1B knockdown increases NB cell sensitivity to cisplatin and reduces invasion and tumourigenic capacity, in association with down-regulation of Notch/Jagged signalling, consistent with a novel NB CSC modulator and therapeutic target[225].
TLX/NR2E1:The orphan nuclear receptor TLX/NRE2 promotes self-renewal in NB CSC-like cells and correlates with poor patient survival. In NB CSC-like cells,TLX/NRE2 is co-expressed with the neural SC/progenitor markers Nestin, CD133 and Oct 4, the neural SC/progenitor migration markers CD15 and MMP-2, and binds and activates Oct4 and MMP-2 promoters[226].
Additional molecular protagonists
MMPs and TIMPS:MMP-9 is involved in almost all of the hallmarks of cancer,including the metastatic behaviour of CSC-like cells[227]. CSC-like I-type but not Stype NB cells exhibit constitutive MMP-9 expression and spontaneous S-type to I-type NB cell CSC-like EMT is associated with NF-κB-induced MMP-9 expression and enhanced MMP-9-dependent invasive capacityin vitro, implicating MMP-9 in NB CSC metastatic behaviour[93,227]. NB CSC-like cells also constitutively express the polycomb transcriptional repressor Bmi1[193], which promotes IkBα degradation,activates NF-κB[191] and increases tumour aggressivenessviathe NF-κB/MMP-9 pathway[188,189]. Bcl2, which also exhibits enhanced expression in NB CSCs, also promotes MMP-9 expression by enhancing NF-κB activity[228], NB cells exhibit methylation-dependent repression of the metalloproteinase inhibitor TIMP-2 and secrete redox enzymes that inhibit TIMP activity, adding to an aberrant MMP/TIMP equilibrium in NB CSCs[210]. TLX/NR2E1-induced MMP-2 expression has also been implicated in the migratory and invasive behaviour of NB CSC-like cells[226].
Nestin:The neural stem/progenitor cell marker and intermediate filament protein nestin is considered to be a selective marker of bone marrow-derived mesenchymal SCs (MSC)[229] and CSCs[230]. Nestin is expressed in NBs, NB CSC-like subpopulations and is associated with aggressive NB behaviour[152,231]. Nestin expression is involved in SC, MSC and CSC self-renewal, angiogenesis and is promoted by Notch signalling, TrkAIII and N-Myc, identifying nestin as a NB-associated, oncogeneregulated, stemness gene[152,229,231].
SPDYA/SPY1:The cell cycle regulatory protein SPDYA/Spy1 controls CDK2 activity at the G1/S transition phase of the cell cycle and is a critical regulator of NB CSC-like behaviour. SPDYA/Spy1 regulates the self-renewal capacity of CD133+NB CSC-like subpopulations[232], upregulates multipotency markers, drives NB tumourigenicity by activating cyclin dependent kinases, and its knockdown decreases c-Myc expression and impairs retinoic acid-induced differentiation of NB CSCs[233].
ALDH1 Isoenzymes:ALDHs are NAD(P)+-dependent enzymes that catalyze aldehyde oxidation to carboxylic acids, and are expressed by and active in haematopoietic SCs and NB CSCs[234]. NB cells express all 19 ALDH isoforms and ALDH1A2 and ALDG1A3 isoforms are potential markers of NB CSCs involved in promoting selfrenewal, tumour sphere growth, colony formation, NB xenografts tumourigenicity and resistance to 13-cis-RA-induced differentiation[235,236].
LIN28B:The RNA binding protein LIN28B is also considered an NB oncogene. Lin28B represses the biogenesis of miRNAs let-7, miR-107, miR-143 and miR-200c by preventing miRNA maturation[237], and is regulated by N-Myc in NB cellsviaan NMyc/miR-26a-5p/LIN28B regulatory axis, and direct binding and activation of the LIN28B promoter[238]. LIN28B binds and activates gene promoters by interacting with the zinc finger transcription factor ZNF143, activating downstream targets that regulate the core regulatory circuitry that controls the malignant state of NB cells, in addition to regulating Gsk3β and L1-CAM expression, involved in cell adhesion and migration[239].
miRNAs, long non-coding RNAs and endogenous retroviruses:Let-7 was the first suppressor miRNA reported to down-regulate tumour formation and metastasis[240],is involved in trunk NC cell migration, is regulated by LIN28B and is involved in LIN28B-mediated trunk sympathoadrenal precursor transformation, supporting roles for let-7 in NB initiation and metastatic potential[241]. Other miRNAs that are altered in NB, include the up-regulated miRNAs: mir-17-92 that targets DKK3, CDKN1A,BIM, MEF2D and ESR1; miR-21 that targets PTEN; miR128 that targets truncated TrkC and up-regulates Bcl-2 expression; miR-380-5p that targets p53; miR-558 that targets HPSE and miR-375 that targets ELAVL4. Down-regulated miRNAs, other than let-7,that are associated with NB include: miR-9 that targets MMP-14; miR-15 that targets Bcl2; miR-103 and miR-107 that target CDK5R1; miR-124 that targets Sox9 and PTPB1;miR-29a that targets cdk6 and CDC6; miR-152 that targets DNMT1; miR-125b that targets TrkC; miR-204 that targets Bcl2 and TrkB; miR-363 that targets ADAM15 and MYO1B and miR-335 that targets Sox4 and TNC. Embryonic SC-associated miR-380-5p cooperates with Ras in primary cell transformation and tumourigenesis, blocks cellular senescence in mice, correlates with N-Myc amplification and poor outcome in NBs and miR-380-5p inhibition promotes p53-dependent NB cell apoptosis and reduces tumourigenesis in an orthotopic mouse NB model[242-245]. A NB CSC suppressor role has been proposed for miR-128, not expressed in primary NBs, which represses the expression of Bmi1 and E2F3a pro-growth transcription factor, and reduces NB cell motility and invasiveness[246-249].
Long non-coding (lnc) RNAs have also been shown to promote and maintain CSCs,and regulate CSC behaviour[250]. LncRNAs implicated in NB CSCs[251-253], include the hypoxia-regulated lncRNA HOTAIR, up-regulated in NBs, which is associated with poor prognosis and CSC self-renewal, differentiation inhibition and high-level PRAF2, which promotes NB CSC proliferation and migration[254]. The hypoxia and N-Myc-regulated lncRNA MALAT-1 also contributes to CSC maintenance[255],promotes NB cell migration, invasive and angiogenic behaviour, and is induced by the N-Myc-regulated histone demethylase JMJD1A, which is also a hypoxia-inducible epigenetic regulator of SC behaviour, providing a lncRNA-mediated mechanism through which N-Myc can promote invasive, angiogenic and metastatic behaviour of NB CSCs[252,256,257]. Repression or polymorphisms in the epigenetic regulator H19 lncRNA have also been implicated in NB CSCs[251], and depletion of lncRNA NBAT1,implicated in retinoic acid-induced NB cell neuronal differentiation, prevents differentiation, and is repressed in undifferentiated CSCs in high-risk N-Myc amplified and non-amplified NB[252,258]. Human endogenous retroviruses (HERVs) have also been implicated in aggressive metastatic behaviour in a variety of tumours. HERV-K activation expands and maintains CD133+CSC-like populations in NC-derived melanoma[259,260], HERV-H lncRNA is a specific marker of pluripotency in human SCs, that interacts with Oct4 to maintain embryonic SC gene expression[259,260] and HERV expression is up-regulated in NB cells by hypoxia and hypoxia mimics[261,262]. Although, no reports link HERV-K or HER-V to NB, it is likely that HERVs also play important roles in promoting and maintaining NB CSCs. Dysregulated HERV-W expression has, however, been reported in NB cells that express the HERV-W trace protein syncitin-1, which exhibits anti-apoptotic activity, promotes immune suppression, and induces EMT, invasion and metastasis, consistent with CSC promoting function. Furthermore, HERV-W induces c-AMP signalling in NB cells,resulting in activation of the syncitin-1 transcription factor CREB[263].
Metabolism, nuclear factor-erythroid 2-related factor 2 and redox regulators:CSCs exhibit unusual metabolism in order to maintain stemness and exhibit anabolic and catabolic metabolic flexibility, associated with alterations in signalling and redox activity, in response to changes within the tumour microenvironment. Normal SCs and CSCs exhibit enhanced glycolytic metabolism that promotes and maintains CSC traits through enhanced expression of glycolysis-related GLUTs, monocarboxylate transporters, hexokinases and pyruvate dehydrogenase kinase 1, the functional inhibition of which reduces tumourigenicityin vivo. Many of these genes are HIFregulated, helping to explain CSC association with hypoxic tumour niches and pseudo-hypoxic states[264,265]. Glycolytic reprogramming in CSCs promotes metastatic progression, enhances therapeutic resistance and involves Snai1 suppression of phosphofructokinase, which switches metabolism from oxidative phosphorylation to glycolysis and promotes EMT. Oxidative phosphorylation is also activated in CSCs, in association with increased mitochondrial numbers and size. This metabolic flexibility is regulated by activated oncogenes, oncosuppressor inactivation,tumour cellular and chemical microenvironments, and result in co-existence of different CSC metabo-types, some fixed and others that exhibit reversible plasticity and can switch back and forth from glycolysis to oxidative phosphorylation,optimizing exploitation of changing tumour micro-environments[264,265]. TrkAIII expression in NB cells promotes metabolic flexibility within a CSC-like state, and in inactive-form localizes to outer mitochondrial membranes in un-stressed NB cells.Under conditions of ER-stress, inactive mitochondrial-associated TrkAIII exhibits Ca2+-dependent translocation to inner mitochondrial membranes, where it undergoes cleavage-dependent activation, resulting in tyrosine phosphorylation of mitochondrial pyruvate dehydrogenase kinase and a metabolic shift to aerobic glycolysis, implicating mitochondrial TrkAIII in an ER-stress-regulated mechanism for metabolic flexibility,within a CSC context and survival-adapted ER-stress response[72,150].
In order to survive the cytotoxic effects of increased ROS production from metabolic and therapeutic sources, CSCs also possess potent antioxidant systems that scavenge and regulate ROS production, ROS-dependent signalling pathways, ROS-regulated transcription factors and defend against ROS-induced cytotoxicity[266]. The nuclear factor-erythroid 2-related factor 2 (NRF2) transcription factor is a master regulator of cellular redox and metabolic homeostasis in normal SCs and CSCs, and is important for regulating CSC quiescent-pseudo-senescent-dormant states, survival, self-renewal,proliferation and differentiation[265,267]. ROS inactivate repressive Keap1/NRF2 interactions, activating the NRF2-dependent antioxidant system, which up-regulates the expression of the anti-oxidants superoxide dismutase, thioredoxin, thioredoxin reductase, glutathione peroxidase, glutathione reductase, ferritin, nicotinamide adenine dinucleotide phosphate, quinone oxi-reductase, glutamate-cysteine ligase catalytic subunit, glutamate-cysteine ligase modifier subunit and increases expression of the drug-metabolism and efflux genes ALDH and ABC transporters[265,267-269]. In tumours, NRF2 weakens oxidative phosphorylation and induces antioxidant gene expression, reducing ROS to levels optimal for CSCs stemness, self-renewal, proliferation, survival, metastatic progression and therapeutic resistance. NRF2 activity in CSCs is modulated by CD44 through p62[270], regulates Nestin, Bmi1, and Sox 2 expression and in NB cells promotes proliferation and inhibits neuronal differentiation[271,272], implicating NRF2 in fine tuning the metabolic/antioxidant system to ensure appropriate ROS levels for NB CSC maintenance. TrkAIII also regulates free-radical ROS levels in CSC-like NB cells by augmenting SOD2 expression, in a TrkAIII/SOD2 axis that increases resistance to mitochondrial free-radical ROS-induced cell death,within the context of a CSC-like phenotype. In this mechanism, increased mitochondrial SOD2 activity attenuates free-radical ROS production and increases H2O2levels,protecting cells against agents that induce free-radical ROS-induced death. Increased mitochondrial H2O2levels promote malignant progression, genetic instability, sister chromatid exchanges, chromosomal translocation, MMP-expression, MMP-dependent invasion and metastatic dissemination, all of which are enhanced by TrkAIII expression in NB cells, identifying the TrkAIII/SOD2 axis as a novel potential CSC therapeutic target[72,152]. NB cells also secrete components of the thioredoxin/thioredoxin reductase redox system, which is enhanced in CSC states. In the extracellular compartment, thioredoxin and thioredoxin reductase promote NB cell invasive behaviour by inhibiting TIMPs and altering the MMP/TIMP equilibrium[273], and disrupt basement membrane integrity, de-regulating endothelial cell tube formation, and potentially promoting EMT[274].
Additional mechanisms involved in NB CSC formation
Cellular polyploidy:The tumour microenvironment, oncogene activation and oncosuppressor inactivation integrate to promote genomic instability and the formation of polyploid giant cancer cells[275], now considered to be an important source of aneuploid CSCs in tumours, including NBs. Theodor Boveri was the first to hypothesize chromosomal polyploidy and subsequent aneuploidy as a mechanism for malignant tumour formation, describing the origin of tumours from a single primordial cell with “abnormal chromosome constitution”, generated by abnormal stimuli that “might induce simultaneous multiple divisions of the centrosomes”,resulting in “tetraploid cells driven to divide by continuous proliferation“, which“opens the door for the production of sarcomatous or carcinomatous progenitor cells”[276]. Today, cellular polyploidy is no longer considered an exclusively pathological process but also a physiological mechanism for tissue homeostasis, that is conserved and subverted in tumour pathogenesis and progression[277]. Between 0.1%-20% of solid tumour volumes consist of polyploid cancer cells that increase in percentage with malignancy, contributing to tumour initiation, heterogeneity, EMT, invasion,metastasis, therapeutic resistance and the generation of CSC-like cells[278-281].Previously, polyploid cancer cells were presumed to be non-dividing but recent observations confirm that these cells express genes involved in cell cycle regulation, in addition to expressing hypoxia-inducible, stem cell-regulating, chromatin remodelling,and invasion and metastasis promoting genes[278,280-282]. Cellular polyploidy can be achieved by cell fusion, an endocycle in which DNA synthesis and G-phases occur without M-phase or cytokinesis to generate single polyploid cells with giant nuclei that exhibit no mitotic features or by endomitosis caused by abortive mitosis that does not result in chromatid separation or cell division and is followed by reentry into S phase, generating multinucleated cells[282]. Most polyploid cells die but others survive initially in a quiescent G0 pseudo-senescent state but then escape this state to undergo asymmetrical division by blebbing and/or bursting to yield aneuploid progeny that go on to generate novel tumourigenic and therapy-resistant CSC-like cells by mitosis[283]. Polyploid tumour cells are generated by tumour microenvironmental stress, within a context of survival adapted cellular ER-stress responses and immunosuppression[279,280,284,285], and are induced by: gene overexpression and oncogene expression that increase centrosome numbers[149,279,280]; replication stress;genotoxic chemotherapeutic agents that induce abortive cell cycles[286-288], and by loss of tumour suppressor function[279,281,289,290]. Tumour cell polyploidy induced by hypoxia is associated with CSC and EMT gene expression patterns, resulting in a more adaptable, less disruptive phenotype within stressful and hypoxic microenvironments, with multiple gene copies lessening the need for chromosome segregation,promoting survival and facilitating tumour evolution and therapeutic resistance.Polyploid giant cancer cells evolve complex karyotypes and exhibit enhanced tumourigenic activityin vitroandin vivoimplicating tumour cell polyploidy as a basis of aneuploidy[279,281]. Within the somatic context, long term passage of induced polyploid somatic cells eventually leads to transformation and tumourigenesis, in a cellular transformation process characterised by transition from diploid to aneuploid statesviatetraploidy. Polyploid cancer cell survival depends upon repression of p53 function and surviving polyploid cells overcome oncogene-induced senescence by repressing p53 and overexpressing DNA repair genes, to form progeny with oncogenic karyotypes and phenotypes, and progressive chromosomal instability, explaining hyper-diploid karyotypes in many malignant tumours[279-282], including NBs[278,291]. Polyploid giant cancer cells exhibit chemo- and radiotherapeutic resistance that results from quiescent or pseudo-senescent states, infrequent slow cell cycles,increased expression of apoptosis inhibitors, p53 functional repression and enhanced DNA repair mechanisms. Resistant polyploid giant cancer cells follow a defined path of chromosome re-organisation and restructuring into polyploid nuclear states that can separate into secondary nuclei that form secondary progeny in a transformation process[279,281,282]. Secondary daughter cells exhibit anchorage-independent growth,undergo reductive mitotic division and produce smaller near-diploid aneuploid cells that proliferate and are tumourigenic, in a process likened to spores in lower organisms that permit survival in harsh conditions and rapid repopulation later. CSCs formed from polyploid cancer cells express CSC markers and exhibit embryonic-like stemness equivalent to a blastomere stage, form different tumour types, illustrating multipotent plasticity greater than marker-purified CSCs, and express fundamental genetic, cellular and developmental programmes for tumour initiation and progression that integrate with “normal” tumour cellular components[292]. CSC-like cells generated from hypoxia-induced CSC marker positive polyploid giant cancer cells, divide asymmetrically and cycle slowly to form aggressive mesenchymal treatment-resistant tumours[279,280,284].
Centrosome amplification:Theodore Boveri also considered that aberrant centrosome numbers were the driving force behind cellular tetraploidy, aneuploidy and malignant tumour formation[276]. Today, it is well recognized that aberrant centrosome numbers characterise polyploid and aneuploid CSCs in solid tumours, implicating mechanisms that promote centrosome amplification in CSCs formation by way of polypoid and aneuploid states. Centrosome amplification in tumour cells has been attributed to Aurora-A kinase overexpression[286]; drug-induced DNA damage-dependent activation of checkpoint kinases Chk1 and Chk-2[287,288]; enhanced polo kinase 4 expression and activity[289]; loss of BRCA1, APC and p53 function[290-292], and oncogenic alternative TrkAIII splicing[149]. In NB cells, spontaneously activated TrkAIII exhibits MT-dependent retrograde transport to the centrosome, where it phosphorylates Plk4, promoting centrosome amplification, augmenting microtubule polymerization, and increasing polyploid giant and aneuploid NB cell formation,chromosomal instability and sister chromatid exchanges[149,153], implicating hypoxia/stress-induced alternative TrkAIII splicing in promoting NB CSCsviapolyploid giant cancer cell formation. This is supported by TrkAIII expression in neural SCs and CSC-like NB cells, associated with enhanced expression of Nanog,Nestin, Sox2, CD133 and CD117 mRNA, and by TrkAIII prevention of NB cell differentiation, promotion of NB xenograft tumourigenic and metastatic capacity, and therapeutic resistance[72], contrasting with fully spliced TrkA promotion of genomic stability in NB cells[293]. Therapy-resistant polyploid giant cancer cells express enhanced levels of Oct4, Nanog and Sox2, involved in self-renewal and apoptosis evasion, and re-proliferate upon pseudo bi-polar centrosome coalescence, resulting in CSC-like progeny that correlates with oncogene amplification, poor prognosis, posttherapeutic relapse, EMT, invasion, metastasis and therapeutic-resistance[280,281]. The formation of polyploid giant cancer cells is the result of centrosome amplification,therefore, represents a critical source of invasive, metastatic and therapy-resistant NB CSC-like phenotypes (Figure 4).
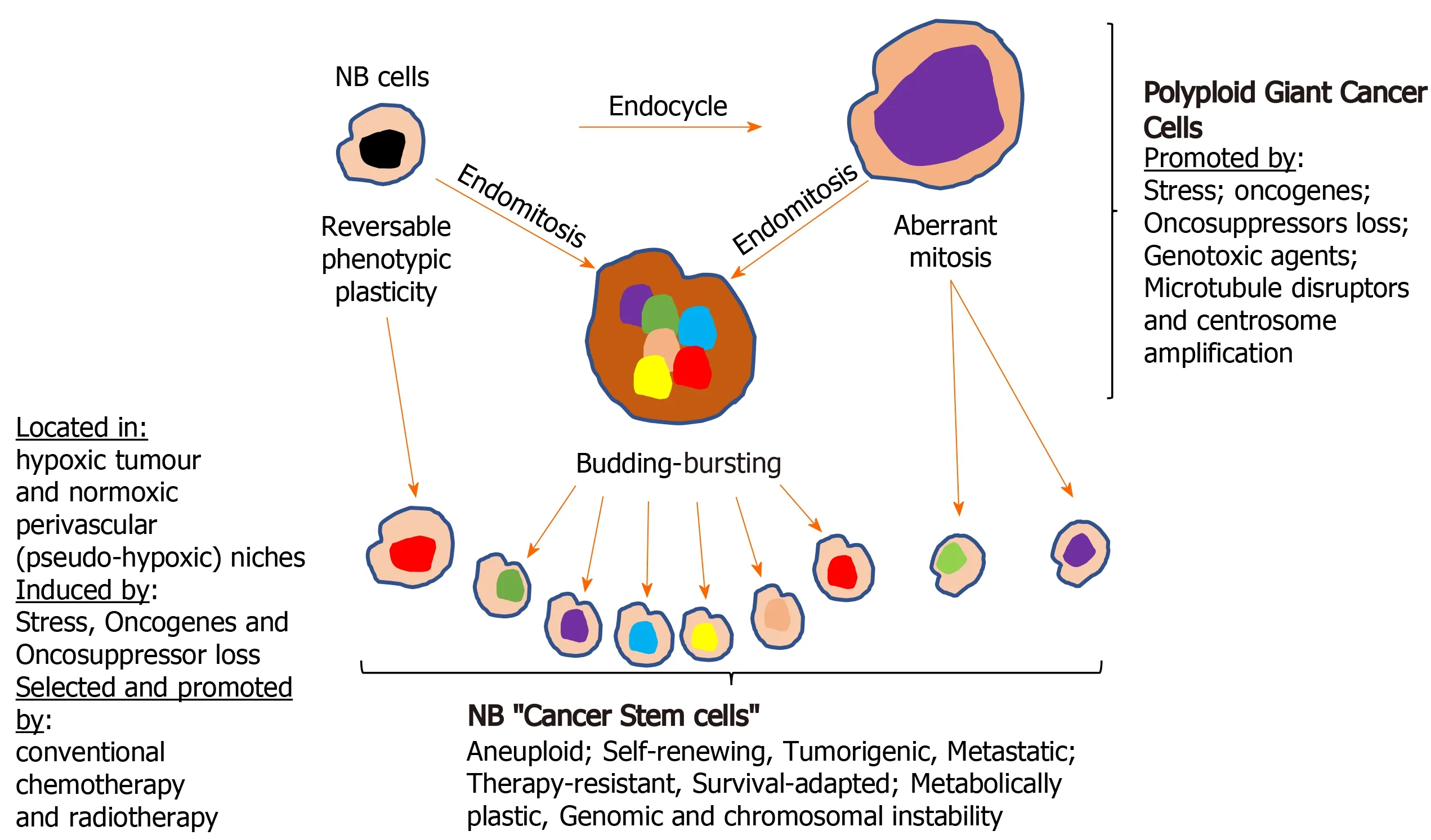
Figure 4 Formation of neuroblastoma cancer stem cells. Illustration of the various way cancer stem cell (CSC)-like cells form in neuroblastomas, by either direct reversible phenotype plasticity from non-cancer stem cells within hypoxic tumour and pseudo-hypoxic perivascular CSC niches and/or via the formation of polyploid giant cancer cells. NB: Neuroblastomas.
Nonsense mediated RNA decay:The multistep nonsense mediated RNA decay(NMD) pathway targets and rapidly destroys both mutated and non-mutated mRNAs,and has been implicated in preventing embryonic SC differentiation[294]. NMD is inhibited by hypoxia in an eIF2a-dependent manner. Stress-induced eIF2a phosphorylation activates stress-response gene expression, resulting in the stabilization of several NMD target mRNAs, stabilized by NMD repression, implicating hypoxia-induced NMD repression in promoting the cellular hypoxic stress-response.Hypoxic repression of NMD involves component re-localization to cytoplasmic stress granules and dynamically alters hypoxic gene expression, consistent with a potential role for NMD repression in hypoxic promotion of NB CSCs[295]. NMD is also repressed by c-Myc, stabilizing Myc target gene expression[296], implicating NMD repression in N-Myc/c-Myc cooperation, which regulates NB CSC phenotypes[201].An NMD/miRNA-128 regulatory circuit has also been identified in which miR-128 represses NMD by targeting UPF1 and MLN51 NMD factors, and in NBs miR-128 exerts a tumour and CSC repressing role, inhibiting expression of the CSC self-renewal factor Bmi1 and the growth promoting transcription factor E2F3a, reducing NB cell motility and invasiveness[246-249].
The stressful hypoxic tumour microenvironment:Hypoxia is involved in NB initiation and progression and promotes NB CSC-like phenotypes[297]. The stressful tumour microenvironment continually selects survival-adapted tumour and tumourassociated “normal” cell populations, resulting in heterogeneous niches within highly aberrant vasculatures that promote episodes of hypoxic, nutrient, metabolic and redox stress. Oxygen pressure is lower in solid tumours than in normal tissues, hypoxic tumours are more aggressive than oxygenated tumours and tumour hypoxia is a common feature of solid tumours, including NB, and represents an independent prognostic marker for disease progression and poor clinical outcome[298-301].Tumour hypoxia, oncogene activation and oncosuppressor inactivation integrate to promote tumour glycolytic metabolic adaptation, resulting in a reducing acidic tumour microenvironment that promotes further adaptation, EMT and progressive dedifferentiation to CSC-like states with enhanced migratory, invasive, metastatic capacity, increased therapeutic resistance and augmented genomic instability[302-305]. In NB, hypoxia promotes more aggressive behaviour that is associated with CSClike subpopulations that localize to hypoxic necrotic areas[306-308]. NB CSC-like cells exhibiting a pseudo-hypoxic phenotype also localize to oxygenated tumour perivascular niches[301,309]. Hypoxia inducible HIF-1α and HIF-2α transcription factors promote metastatic CSC-like phenotypes[310,311], share target genes but also exhibit target specificity and differences in regulation. HIF-1α proteins are stabilized by hypoxia but HIF-1α transcription is not induced by hypoxia, whereas hypoxia induces HIF-2α transcription. HIF-1α is involved in acute hypoxic responses and subsequently decreases, whereas HIF-2α function is maintained during prolonged hypoxia. Hypoxia-independent HIF expression is promoted by growth factor signalling, including IGF, VEGF and EGF signalling through the PI3K and Ras/MAPK pathways. In NBs, HIF-2α expression correlates with aggressive disease, HIF-2α+/HIF-1α-mesenchymal CSC-like NB cells populate perivascular NB regions and NB cells cultured at 5% O2to mimic end capillary O2diffusion levels, express HIF-2α but not HIF-1α, implicating HIF-2α in pseudo-hypoxic NB CSC-like phenotypes within perivascular NB areas, potentially driven by autocrine GF/GFR or oncogene-dependent signalling[301,309,312]. HIF-2α is also implicated in delta like noncanonical notch ligand 1 (DLK1) promotion of NB CSC-like phenotypes. DLK1 is regulated by HIF-1α and HIF-2α, induced by hypoxia, is a bona fide neural SC marker required for selfrenewal[117,181] and is robustly expressed by undifferentiated tumour cells. In NB xenografts, DLK1+cells preferentially localize to hypoxic areas[116,117], and together with HIF-2α promote aggressive tumour behaviour and undifferentiated CSC-like states[301,309,312]. HIF-1α also promotes NB CSC-like phenotypes within the context of an aberrant unfolded protein response, characterised by reduced ATF3 expression.In this mechanism, HIF-1α induces Id1 expression in NB cells that do not express ATF3, which down-regulates Id1 expression in most cell types[295]. Intermittent hypoxia also promotes NB CSC-like phenotypes involving HIF-1α and HIF-2α expression, resulting in enhanced survival and clonogenic activity, increased Oct4,CD133, Id2, Hes1, c-kit and Notch-1 expression, reduced expression of SNS differentiation markers NPY, Hash1 and dHand, and HIF-1α knockdown promotes neuronal differentiation-associated NeuN and NF-M expression[313]. Hypoxia-induced alternative splicing also promotes CSC-like phenotypes and EMT by promoting HIF-1 α interaction with Zeb1 and a soluble sCD44 splice variant, induces DCLK1 alternative splicing to promote CSC self-renewal and chemoresistance, and promotes alternative TrkAIII splicing in NB cells, enhancing angiogenic, tumourigenic and metastatic behaviour associated with the context of a more CSC-like phenotype[314].
Mesenchymal stem cells:MSCS invade NB tumour tissues[315] and NB cells that express CXCR4 and CXCR7 chemokine receptors migrate towards MSCs that express SDF-1 in the bone marrow[316]. MSCs isolated from NBs promote tumourigenesis by interacting with NB and stromal cells, and are more frequently arrested in G0/G1 phases of the cell cycle, facilitating therapeutic-resistance[317]. Normal MSCs therefore are NB stromal components that regulate NB CSC tumourigenic and metastatic behaviour.
THERAPEUTIC PROSPECTS FOR TARGETING AND ELIMINATING NB CSC-LIKE POPULATIONS
Considering the young age of NB patients, the aggressive nature of the disease and the concept that post-therapeutic disease-relapse, metastatic progression and eventual mortality result primarily from the selection of therapy-resistant CSC-like cells, it is of paramount importance to develop novel therapeutic strategies that target and eliminate NB CSC subpopulations in order to improve therapeutic response-duration and survival.
CSC-like NB subpopulations reflect not only the primitive embryonic origins of this tumour but also evolutionary processes promoted by accumulating genetic changes within a continuously evolving tumour microenvironment, also altered by conventional therapeutic strategies, which together select survival-adapted CSC-like phenotypes. CSC therapeutic resistance depends upon numerous mechanisms,including an intermittent slow cell cycle, enhanced expression of drug efflux ABC transporter and P-glycoprotein, enhanced expression of apoptosis inhibitors and repression of p53 function, a flexible pro-glycolytic metabo-type associated with highlevel anti-oxidant expression, enhanced DNA repair mechanisms, survival-adapted ER-stress responses, formation of polyploid giant cancer cells, CSC localization to hypoxic niches, the present physical tumour barriers to therapy, and phenotypic plasticity, permitting CSC regeneration from non-CSC tumour cells[280,318-320].These factors combined with CSC similarities to normal SCs, make specific therapeutic targeting and elimination of NB CSC populations a daunting problem.
In this next section, we review novel potential therapeutic strategies, currently aimed at targeting and eliminating NB CSC populations or that could be adapted for use in NB.
Targeting cell surface and membrane-associated molecular protagonists
CSC cell surface marker targeting with antibodies and CAR cell strategies:CD44 was the first CSC marker to be targeted with monoclonal antibodies in acute myeloid leukemia, with successful disease eradication[321], suggesting that CD44 in an aggressive NB CSC-like subpopulation could also be targeted[112]. Targeting CD133,expressed by NB CSC-like cells[92], with antibodies conjugated to the cytotoxic agent monomethyl-auristatin has been reported to be internalized and to kill CD133+liver cancer CSCs[322], and targeting CD144 with antibodies, or targeting STAT3, in CD114+NB CSCs that exhibit self-renewal and high tumourigenic activity, depletes the NB CSC subpopulations, decreases tumourigenicity, reduces metastatic capacity and increases chemotherapeutic sensitivity[323].
CAR-T strategies eradicate tumour cells by recognizing cell surface antigens and,unlike natural T-cell receptor positive T cells, recognize antigens by an MHCindependent mechanism. CARs are comprised of an extracellular single chain variable tumour antigen-specific fragment linked to extracellular spacer, transmembrane and intracellular signalling domains. Latest generation CAR T-cells express inflammatory cytokines and can effectively and specifically eliminate CSCs. Pre-requisites for CSC CAR-T therapy include cell surface expression of CSC-specific, preferably functional,tumour antigens. CAR T-cells that specifically recognize CSC markers GD2, LGR5,IGFR-1, CD44, CD47, EpCAM, Dll4, FZD and CD123 have been generated, do not promote CSC enrichment and do not exhibit substantial tissue cytotoxicity, and CAR T-cells that recognize CD133, CD166, CD20, CD38, CLL-1, EpCAM, CD123, CD171,ROR1, CD44, CD47, CD117 and c-Met are presently being evaluated in clinical studies[159]. For NB therapy, targeting L1-CAM (CD171), which drives NB CSC invasive and migratory capacity, with L1-CAM CE7 epitope CAR T-cells developed for NB immunotherapy, is safe and efficacious in refractory recurrent NB, and CAR T-cells that target the CE7 epitope of CD171 (CE7-CAR-T cells), generated from NB patients with recurrent/refractory disease, has also been shown to be safe and efficacious[324].
Potential problems with CAR-T therapy include off-target toxicity due to low-level antigen expression in normal tissues. This can be overcome using CAR T-cells that recognize two or more antigens, enhancing tumour specificity, or CAR T-cells that express an inhibitory receptor against normal antigens in addition to recognizing a tumour specific antigen. Alternatively, considering that CAR antigen affinity is key to activating cytotoxicity, CAR T-cells with low affinity CARs that are only cytotoxic when they bind high density antigens may minimize off-target side effects, and CAR T-cells armed with caspase-9, sensitive to AP1903-induced activation, can ensure CAR T-cell elimination under conditions of aberrant cytotoxicity[325]. CAR T-cells engineered to contain a lenalidomide-inducible dimerization system that serves as an ON- and OFF-switch, characterised by lenalidomide-dependent anti-tumour activity in the presence of tumour antigen and lenalidomide-induced degradation in the absence, may further minimize potential off-target side effects[326]. Other problems associated with CSC-targeted therapy, include phenotypic plasticity, illustrated by reversible bidirectional conversion between CSC and non-CSC states induced by changes in the tumour microenvironment and therapeutic agents, which may render target-therapies, including CAR T-cells only transiently efficacious, until novel CSClike subpopulations regenerate. In this case, efficacy could be maximized by combining CAR T-cell therapy with conventional chemo and/or radiotherapy therapy to kill both CSC and non-CSC tumour cells, supported by a report of synergistic efficacy in a rat glioma model, when NKG2D CAR-T therapy is combined with conventional radiotherapy[327], and when EpCAM CAR-NK92 CAR-T therapy is combined with regofinib plus salinomycin, metformin and S-phase kinase associated protein 2 inhibitors and low dose conventional chemotherapy[328,329]. Heterogeneity of CSC tumour antigen expression can also be mitigated using bi- or poly-specific CAR T-cells that target multiple tumour antigens, and CSC tumour antigen expression could be promoted to facilitate CAR T-cell recognition, exemplified by retinoic acid up-regulation of CD38 expression that sensitizes acute myeloid leukemia cells to CD38 CAR T-cells[330]. CAR T-cell exhaustion and senescence can be overcome using 4-IBB and ICOS co-stimulatory domains, CAR T-cell tumour infiltration and durability can be enhanced, and exhaustion and senescence reduced, combined with immune checkpoint PD-1, CTLA4, TIM-3, LAG-3 and adenosine 2A receptor inhibitors. CAR Tcells that express anti-apoptotic factors can reduce tumour-induced CAR T-cell apoptosis, CAR T-cells that express interleukin (IL)-7 and IL-15 can improve persistence, and CAR T-cells that express chemokine receptors improve tumour infiltration and homing to CSC niches, as does local or intra-tumour CAR T-cell delivery[159]. Physical tumour stromal barriers can be disrupted by CAR T-cell expressing extracellular matrix degrading enzymes targeted to tumour stromal FAP+fibroblasts[331], and tumour oxygen supplementation can enhance infiltration and reduce immunosuppression[332]. Repression of CAR-T function by tumour-associated Tregulators, macrophages, neutrophils, myeloid-derived suppressor cells and inhibitory IL10, TGFβ, PGE2, arginase, cyclooxygenase-1, galectin-9 and idoleamine 2, 3 dioxygenase molecules, can be reduced using specific inhibitors[159]. Other modifications that may improve efficacy include adjuvant tumour de-bulking prior to CAR Tcell therapy to reduce toxicity due to cytokines and adjuvant chemotherapy with agents that induce T cell chemokine expression can pre-condition tumours for CAR Tcell therapy. Further characterisation of CSC-specific targets can reduce the risk of autoimmunity and improved understanding of how specific CSC antigens alter during disease progression will help fine-tune individualized CAR T-cell therapeutic approaches, which combined with targeting monoclonal antibodies, conventional chemotherapy and radiotherapy, unveil an important therapeutic future for CAR Tcell based immunotherapies in the eradication of NB CSCs.
Invariant natural killer T cells (INKTs) or natural killer cells (NKs) represent an alternative to CAR T-cell therapy, with advantages including potential clearing of tumour associated macrophages and better penetration. INKTs alter the behaviour of tumour-associated macrophages and myeloid derived suppressor cells, secrete cytokines that recruit immune effectors and kill neuroblasts and CSCs[333]. INKTs stimulated with a-galactosylceramide (GAg), adoptive iNKT transfer and CAR-iNKTs could all be utilized to target NB CSCs. GAg-stimulated iNKTs enhance NK-mediated NB cell killing, reinvigorate exhausted CD8+ T cells[334,335] and show good tumour infiltration when combined with immune checkpoint inhibitors. Adoptive iNKT transfer in humanized mice reprogrammes NB xenograft-associated macrophages from the M2 to the anti-tumour M1 phenotype[336], GD-2 specific CAR-iNKTs reduce NB xenograft growth in mice and prolong survival, and CAR-iNKTs are being evaluated in phase I clinical trials in relapsed or refractory NB[333]. With respect to NKs, Dinutuximab an anti-GD2 antibody was the first immunotherapy used for NB,and exerts its effects also by stimulating NK activity, implicating a potential use for NK activators, including novel bi- and tri-specific killer cell engagers that enhance NK cytotoxicity, in NB therapy. Alternatively, autologous adoptive transfer of activated NK cells combined with anti-GD2 antibodies could be used, which have been reported to increase survival in a mouse NB xenograft model[337]. Along the same theme,allogenic NK transfer combined with humanized anti-3F8 antibody and IL-2 is currently in clinical trial for NB[338] (NCT 02650648) and CAR-NKs evaluated in NB include GD2-CAR-NKs that exhibit specific robust GD2+NB cell killing and NKG2D CAR-NKs that kill NB-associated myeloid derived suppressors[333].
Inhibition of immune checkpoint protagonists PD-1, CTLA4, TIM-3, LAG-3 and adenosine 2A receptor, and in particular PD-1/PD-L1 and CTLA4, also represents an important approach for restoring anti-tumour immunity and improving CAR T-cell,CAR-iNKT, CAR-NK, iNKT and NK immunotherapeutic approaches[339]. N-Myc amplified NBs exhibit high-level PD-L1 expression, which correlates with reduced tumour infiltrating T-cells and unfavourable prognosis, and can be reduced by shRNA-mediated functional N-Myc inhibition or the BET bromodomain inhibitor JQ1[340].
ABC and P-glycoprotein transporter inhibitors:ABC transporter inhibitors in general lack specificity and elicit important toxic side effects[123]. However, the ABC inhibitor probenecid sensitizes NB CSCs to cisplatin, increasing apoptosis and decreasing proliferation associated with caspase 3 expression, reduced ABC transporters MDR1, MDR2 and BCRP expression and reduced expression of CD133 and vimentin and snai1 EMT markers, forcing CSCs into a non-CSC state, associated with therapeutic re-sensitization[341]. The prophylactic antianginal agent Perhexiline induces non-coding Alu RNA neuroblastoma differentiation marker 29 expression in NB cells, represses ABC transporter expression, increases sensitivity to cisplatin, depletes CSCs through differentiation, reduces NB nodular formation and enhances survival in a mouse NB xenograft model[342]. The P-glycoprotein inhibitor vardenafil inhibits P-glycoproteinmediated drug-efflux and increases the cytotoxicity of paclitaxel and vincristine in enriched CSCs[343].
Inhibiting oncogenes and signalling pathways
NB CSC-associated oncogenes:Small RNA inhibitors that target E6 mRNA in CSCenriched cervical cancers[344] and HMGA1 in glioblastoma CSCs[345] are proof of concept the NB CSC-associated oncogenes could be silenced. However, delivery of high enough concentrations of oncogene-specific small RNA inhibitors (antisense oligonucleotides, RNA interference, ribozymes) to tumours is a significant clinical problem, which may be overcome using nanoparticles, liposomes and PEGylated aptamers (see below) targeted to cell surface CSC markers (see above)[346]. NB CSCassociated Alk, TrkB and TrkAIII oncogenes could be inhibited by the Food and Drug Administration (FDA)-approved small molecule ALK inhibitors Crizotinib, Ceritinib,Alectinib, Brigatinib, Entrectinib and Lorlatinib[347] or by the FDA-approved small molecule Trk kinase inhibitors Entrectinib, Larotrectinib, Lestaurtinib, GNF-4256 or GNF-5837 either alone or in combination with conventional chemotherapy[72,348-350], with the drug-resistance evolution counteracted by parallel development of modified drugs specific for known and expected kinase-domain mutations, and efficacy enhanced when combined with conventional chemotherapy and/or radiotherapy. Alk, TrkB and TrkAIII oncoproteins could also be reduced using appropriately CSC-targeted delivery of gene-specific peptide nucleic acids or siRNAs[72]. Oncogenic alternative TrkAIII splicing could be reversed using new generation lentiviral vectors that express TrkAIII inhibitory siRNAs and full length fully-spliced TrkA complementary DNA, mutated to prevent inhibition of expression without altering the amino acid sequence, in order to block TrkAIII oncogenic activity and reinstate TrkA tumour-suppressing activity[72]. Ibrutinib and acalabrutinib inhibitors of Bruton’s Tyrosine kinase, which is highly expressed in N-Myc amplified NBs and is associated with poor prognosis, have been shown to inhibit the migratory, invasive and tumour sphere forming capacity of NB cell lines, and to synergize with cisplatin,identifying Bruton’s Tyrosine kinase as a potential druggable NB driver oncogene, of relevance to NB CSCs[351]. Survivin, an oncogenic inhibitor of apoptotic and autophagic cell-death, is also differentially expressed in CSCs, is associated with chromosome 17q gain and adverse outcome[352], and can be inhibited by a significant number of small molecule and mRNA inhibitors (antisense oligonucleotides, RNA interference, ribozymes)[353].
CSC-associated signalling pathways:CSC signalling pathway inhibitors could also be adapted for use in NB, including the Notch pathway inhibitor Psoralidin, which reduces tumour-sphere formation, induces apoptosis, inhibits proliferation and increases doxorubicin and docetaxel sensitivity in breast cancer CSCs[354]. Shh signalling is also essential for NB cell proliferation and tumourigenicity[355]. The Shh signalling antagonists cyclopamine, IPI-926 and GANT61 promote tumour regression and deplete CSC in cancers, including NB[356-359], decrease CD133 and CD15 double positive NB CSC-like cells and reduce NB tumourigenic activityin vitro[360]. Small molecule Wnt and CD44 signalling inhibitors that reduce breast cancer CSC selfrenewal[361,362] and the cyclin dependent kinase inhibitor Roniciclib (BAY 1000394)that also targets CSCs, exhibit potent therapeutic effects in high-risk NB, by inhibiting Wnt/β-catenin signalling, inducing nucleolar-stress, down-regulating CSCs and inhibiting proliferation[363,364]. The Wnt/β-catenin signalling inhibitor Pimozide also blocks CSC EMT, inhibits CSC-associated STAT-3 and represses differentiation inhibiting gene expression[365]. Other inhibitors include, the irreversible Glycogen synthase kinase-3b serine/threonine kinase inhibitor Tideglusib, which reduces NB cell proliferation, migration and viability by eradicating the self-renewal capacity of highly resistant NB CSCs, reducing tumour sphere forming capacity and CD133 expression[366]. The AKT/mammalian target of rapamycin (mTOR) signalling inhibitors triciribine and rapamycin target NB CSCs, decreasing survival and tumour sphere-forming capacity[367], and the mTOR inhibitor temsirolimus, which combined with the P53 activating MDM inhibitors Nutlin 3a or RG7388, inhibits proliferation and induces apoptosis inin vivoNB models[368]. Dual inhibition of the AKT2/mTOR and MAPK pathways with specific CCT128930 AKT2 inhibitor and PD98059 MAPK inhibitor decreases NB CSC proliferation, migration, tumour sphere-forming and angiogenic capacity[369]. Retinoic acid combined with Dlk1 knockdown disrupts CSCs by reducing DLK1 interactions with PHB1 and PHB2 prohibitins, required for selfrenewal and clonogenic capacity[118,370]. Targeting STAT3/JAK signalling with the natural polyphenol curcumin enhances NB chemosensitivity[371]. The highly oxidized plant steroid cucurbitacin-I either alone or in combination with the bi-phenol Honokiol inhibits NB tumourigenicity[372,373]. The selective Aurora B kinase inhibitor AZD1152 reduces NB cell proliferation and is selectively cytotoxic to tumour inducing NB CSC-like cells[374]. Inhibition of adenosine monophosphate activated protein kinase (AMPK) signalling also targets CSCs, the AMPK activator Metformin and AMPK pathway inhibitor vidarabine or 9-β-D-arabinofuranosyladenine reduce 2-D and 3-D NB CSC proliferationviaMetformin inhibition of mTOR and Akt, and energy deprivationviaAra-a-mediated AMPK inhibition[375]. The PI3K/mTOR inhibitor NVP-BEZ235 decreases angiogenesis and increases survival in N-Myc-amplified orthotopic NB xenograft and transgenic N-Myc driven mouse NB models by reducing CSC-like activity[376].
Inhibition of transcription factors and regulators
N-Myc: NB CSC-associated N-Myc oncogene expression is stabilized and potentiated by interaction with ubiquitin-specific protease 7, and could be inhibited by the ubiquitin-specific protease 7 inhibitor P2207[377]. N-Myc expression in NB cells can also be reduced by small molecule BET bromodomain inhibitors JQ1 and I-BET151,which combined with HDAC inhibitor Panobinostat act synergistically to reduce NMyc and LIN28B CSC-promoting oncogene expression in NB cells, resulting in apoptosis, reduced tumour growth and progression in NB-bearing mice[378].Inhibitors of N-Myc expression would also reduce N-Myc regulated MALAT1 lncRNA which contributes to CSC induction and maintenance[255] and reduces the expression of N-Myc-regulated histone demethylase JMJD1A, a regulator of SC behaviour and CSC-associated MALAT1 expression in N-Myc amplified NB[251,256,257].
HIFs:Tumour hypoxia and pseudo-hypoxic phenotypes drive NB CSC phenotypes by activating HIF-1 and HIF-2 transcription factors, identifying HIFs are potential therapeutic targets in reducing NB CSC subpopulations. HIF inhibitors include inhibitors of HIF mRNA and protein expression, HIF dimerization, DNA-binding and transcriptional function and promoters of HIF-α degradation. HIF-1 mRNA and protein expression inhibitors include: lncRNA PIN1-v2[379]; S-TRPM2 calciumpermeable ion channel short variant[380] and EZN-2698 and EZN-2208 HIF-1α antisense oligonucleotides[381,382]. The topoisomerase inhibitor topecan inhibits HIF-1α protein translation and function[383], the natural flavonoid chrysin, soybean glyceollin phytoalexins and KC7F2 small molecule inhibit HIFα protein expression[384-386], and the estrogen metabolite 2-methoxy-estradiol inhibits HIF-1α and HIF-2α protein synthesis, nuclear translocation and transcriptional activity[387]. Hsp90 inhibitors GA, 17-AAG, 17DMAG and EC154, the HDAC inhibitor vorinostat and LW6t small molecule promote VHL-dependent HIF-α degradation, preventing accumulation and inhibiting transcriptional activity[388-391]. The small molecule PX12 inhibits HIF-1α accumulation by targeting thioredoxin-1[392,393], and BAY87–2243 suppresses HIF-1α and HIF-2α protein accumulation by inhibiting mitochondrial complex-1 (stopped in phase 1 trials)[394,395]. Inhibitors of HIF dimerization, include:cyclic CLLFVY that binds the HIF-1α PAS-B domain disrupting dimerization,transcriptional function and tumour cell hypoxic responses[396]; TC-S7009, a nanomolar HIF-2 but not HIF-1 inhibitor that impairs DNA-binding and HIF-2-dependent hypoxic responses[397], and acriflavine that inhibits HIF-1 and HIF-2, and prevents HIF dimerization[398]. Doxorubicin and daurubicin inhibit HIF binding to HREs in gene promoters[399] and echinomycin (NSC-13502) prevents HIF-1 binding to core 5′-CGTG-3′ HRE sequences in the VEGF promoter[400]. HIF transcriptional inhibitors,include: chetomin dithio-diketopiperizine that impedes HIF-1α interaction with its transcriptional activating histone acetyltransferase p300 co-factor and increases radiosensitivity[401]; idenopyrasole 21 that inhibits HIF-1 transcriptional activity[402];the platelet aggregation inhibitor YC-1 that disassociates HIF-1α/p300 complexes,represses HIF transcriptional activity and reduces HIF-1α protein accumulation[403];FM19G11 that inhibits HIF transcriptional activation by inhibiting interaction with p300[404]; NSC-607097 small molecule that inhibits HIF-1 transcriptional activity[405]and IDF-11774 that prevents HIF-1α accumulation, regulates cancer metabolism and suppresses tumour growthin vitroandin vivo[406]. Chaetocin, a de-regulator of HIF-1 α pre-mRNA splicing, also inhibits angiogenesis and growth in cancer models[407,408]. HIF inhibitors would also block hypoxia-induced CSC-associated lncRNA expression by inhibiting HIF-induced histone demethylase JMJD1A expression, a regulator of SC behaviour and CSC-associated lncRNA MALAT1 expression in NB[251,256,257].
HIF-1α silencing, when combined with retinoic acid formulations such as Al trans retinoic acid, used in the clinic to induce differentiation in high-risk NB following induction and consolidation chemotherapy[409,410], augment NB cell differentiation,senescence and chemosensitivity[411], and when combined with the proteasome inhibitor MG132 reduce NB CSC tumour sphere forming capacity, Nestin, Sox2 and Oct4 expression[412], highlighting the importance of combining HIF inhibitors with differentiation inducers to target malignant NB CSC subpopulations.
Targeting epigenetic transcription regulators:Targeting DNA methylation machineries with DNA methylase inhibitors has also been proposed as a potential therapeutic strategy for eliminating CSCs and their progenitor cells[413]. The histone deacetylase inhibitor vorinostat enhances NB cell chemosensitivity, inhibits NB CSC tumour sphere formation, depletes SP CSC-like NB cells and reduces invasion, in association with altered expression of the CSC markers ABCB1, ABCC4, LMO2, SOX2, ERCC5,S100A10, IGFBP3, TCF3, and VIM and drug-resistance genes[414]. The HDAC inhibitor CBHA, combined with retinoic acid, exerts a synergistic anti-tumour effect in a human xenograft NB models by inhibiting survivin expression and AKT signalling[415,416]. HDAC inhibitors combined with Al trans retinoic acid reduce NB cell proliferation[417]. SiRNA inhibition of the polycomb repressor complex component Bmi1 restores NB cell expression of Kif1Bβ tumour suppressor, inducing apoptosis and promoting NGF/TrkA-dependent NB cell differentiation[70,192]. The HDAC inhibitor Panobinostat, combined with BET bromodomain inhibitors, reduces N-Myc and Lin28B oncogene expression in NB cells, inducing apoptosis, reducing proliferation and blocking tumourigenicity in a mouse NB model[378]. Methylation silencing of TIMP-2 in NB cells is reversed by methyltransferase inhibitors[210], methylation silencing of caspase-8[418] can be reversed by methyltransferase inhibitors, and combined with cisplatin or etoposide promotes apoptosis[419], and with INFg facilitates TRAIL-induced apoptosis[420]. DNA methylation inhibitors, however, also promote NB CSC gene expression and phenotypes, and within the context of NB promote completely undifferentiated large cell NBs, the most aggressive and deadly form of NB[421]. The histone demethylase JMJD1A inhibitor DMOG has been reported to reduce CSC-associated lncRNA MALAT1 expression in N-Myc amplified NB[256].
Inhibiting redox enzymes
Consistent with enhanced anti-oxidant activity in CSCs and potential extracellular roles for Trx and TrxR in altering the MMP/TIMP balance, disrupting extracellular matrices, promoting invasion and altering the angiogenic behaviour of endothelial cells[273,274], novel TrxR1 inhibitors have been reported to induce CSC death,suppress multidrug resistance by inhibiting the P-glycoprotein extrusion pump, to inhibit antioxidant defense, suppress invasion and migration and enhance sensitivity to chemotherapy[421]. Increased mitochondrial SOD2 expression and activity in NB CSCs, promoted by oncogenes, including TrkAIII, also enhances NB cell resistance to ROS-induced cytotoxicity[152], and could be targeted to enhance chemosensitivity. In this regard, the nonsteroidal anti-inflammatory drug Diclofenac has been shown to inhibit mitochondrial SOD2 expression and activity, and to promote NB cell apoptosis through the intrinsic mitochondrial pathway[422].
Alternative CSC inhibitors
Other agents reported to target CSCs, include the plant alkaloid Berberine, which attenuates CD133, β-catenin, N-Myc, Sox2, Notch-2 and Nestin expression in Neuro2A NB cells impairing cancer stemness; down regulates PI3K/Akt and Ras/MAPK/Erk signalling reversing EMT in association with restored E-cadherin expression and reduced vimentin and fibronectin expression; represses cyclin and cyclin-dependent kinase expression potentiating G0/G1 cell cycle arrest and inhibiting proliferation, and modulates TGFβRI and RII receptors, promoting neuronal differentiation[423]. The polo-like kinase-1 inhibitor imidazotriazine GSK461346 (in clinical development),inhibits clonal expansion, promotes cell death and reduces tumourigenic activity in tumour initiating NB CSC models[424]. The small molecule tankyrase inhibitor XAV939 reverses NB CSC stemness and reduces migration capacity[425]. TRAIL induces apoptosis in TrkAIII expressing CSC-like NB cells through SHP/Src-mediated crosstalk with the TRAIL-receptor signalling pathway, suggesting that improved TRAIL formulation may also have a place in eradicating CSC populations, under certain circumstances[426]. The mitochondrial carnitine palmitoyl-transferase-1 inhibitor perhexiline promotes NB cell differentiation by inducing non-coding RNA NB differentiation marker 29 expression, resulting in reduced stemness, anchoragedependent growth, reduced tumourigenic capacity and enhanced chemosensitivityviareduced ABC transporter expression and, combined with cisplatin, reduces nodule formation and increases survival in a mouse xenograft NB model, consistent with CSC depletion[342,427]. Stem cell-based small molecule screens have identified Dequalinium analogue C14 Linker, an analogue of the antibacterial mouthwash component Deca 10, as a nanomolar cytotoxic agent that kills tumour initiating CSClike NB cells, reduces tumour sphere formation and targets NB CSCsin vivoby impairing mitochondrial function and inducing apoptosis[428]. Rapamycin has also been identified as an inhibitor of NB tumour initiating CSC-like survival and proliferation at nanomolar concentrations, and when combined with vinblastine inhibits NB xenograft tumour growth[428]. Additional compounds that exhibit selective activity against tumour initiating NB CSC-like cells include teniposide and vinblastine[428]. A drug library screen has also identified carbenoxolone as an inhibitor of FOXO-3-dependent therapeutic-resistance in high-stage NB cells, reducing survival and increasing sensitivity to chemotherapeutic agents in 2D and 3D models[429]. bcarotene also inhibits NB CSC self-renewal capacity and reduces CSC marker expression and resensitizes NB CSCs to cisplatin cytotoxicity, suggesting a potential use in NB chemotherapy[430]. Considering that neural-related tumour initiating CSClike cells exhibit distinct telomere maintenance, NB CSC-like cells may also represent good targets for telomerase inhibitors[431].
Targetting polyploid giant cancer cells and centrosome amplification
Polyploid giant cells are therapy-induced, therapy-resistant tumour subpopulations capable of orchestrating post-therapeutic recurrence and disease progression through the generation of CSCs[280,281,283,432]. CSC Polyploidy is considered a druggable phenotype but mitosis targeting strategies may also promote polyploidy in surviving cells. Novel therapeutic strategies include targeting the higher energetic needs of polyploid giant cells[433]. In this regard, brain and acute myeloid leukemia polyploid giant cells are preferentially killed by the glycolysis inhibitor 2-deoxy-D-glucose[434,435]. Specific mTOR inhibitors induce NB cell osteogenic differentiation[436] and enhance the effect of Aurora kinase inhibitors in promoting apoptosis of polyploid giant cancer cells[433]. The inhibition of mTOR signalling prevents therapy-induced polyploid giant cell formation[437] and the mTOR inhibitor AZD8055 prevents therapy-induced formation of pancreatic cancer polyploid giant cancer cells[438],illustrating that metabolic inhibition in polyploid giant cells may not only reduce CSC generation but also enhance therapeutic sensitivity in tumours containing polyploid giant cells. The anti-fungal anti-oxidant resveratrol selectively reduces the fitness of polyploid giant cancer cells by slowing down the cell cycle and promoting apoptosis,an effect that is augmented when combined with AMPK stimulators. In this regard,resveratrol combined with aspirin reduces the formation of polyploid colon cancer cells that lack p53 function and in APCMin/+mice, prevents polyploidy in intestinal cells and subsequent tumour formation[439,440]. Conversely, maintenance of polyploid cancer cells senescence may also be efficacious, illustrated by the small molecule inhibitor R1530, which promotes tubulin polymerization and mitotic checkpoint kinase BubR1 function, inducing senescent polyploid cancer cell formation and reducing tumour growth in a human H460 non-small-cell lung cancer xenograft model, suggesting that continual promotion of mitotic catastrophe in polyploid cancer cells may block NB CSC formation[441]. Along this theme, microtubule polymerizing taxanes and microtubule de-polymerizing vinca alkaloids promote mitotic catastrophe and death in cancer cells but lack specificity and induce severe side-effects; investigational Monastrol AZD4877, Ispinesib, and ARRY-520 (Phase 1 and II trials completed)Kinesin-5 motor protein inhibitors promote mitotic arrest, tumour cell death and are well tolerated; FDA-approved GSK923295 centrosome-associated protein-CENP-E inhibitor induces defective mitosis and inhibits proliferation; FDA-approved UCN-01/staurosporine and AZD7762 check-point kinase inhibitors induce death in p53-deficient tumours; WEE1 kinase and HDAC inhibitors combined with DNA damaging agents induce mitotic catastrophe; APC-Cdc20 targeting prevents cyclin B degradation and promotes mitotic exit; small molecule dynamin GTPase inhibitors induce cytokinesis failure and cell death, and c-myc repression promotes cancer cell mitotic catastrophe and death[442].
The central role of aberrant centrosome numbers and behaviour in polyploid giant cancer cell formation, continuous chromosomal instability, generation of aneuploid CSC-like cells, de-regulated microtubule organisation and irregular cell cycles, also makes the centrosome a promising therapeutic target for reducing tumour CSC populations[443,444]. Centrosomes and their pericentriolar matrices contain important proto-oncogenes and tumour suppressors that, when altered, drive centrosome amplification and subsequent chromosomal aberrations, the therapeutic inhibition of which can blunt centrosome involvement in tumour progression. Targeting centrosome Plk1 with BI2536 or BI6727 inhibitors has shown efficacy in acute myeloid leukemia[445]. Small molecular cyclin-dependent kinase inhibitors that de-regulate the centrosome cycle are in development[446], FDA-approved PD0332991 (Palbociclib),LEE011 (ribociclib) and LY2835219 (abemaciclib) cdk4/6 inhibitors are amongst the most recent[444] but may cause rare severe lung inflammation (https://www.fda.gov/drugs/drug-safety-and-availability/fda-warns-about-rare-severe-lunginflammation-ibrance-kisqali-and-verzenio-breast-cancer). A number of Aurora kinase inhibitors are also in clinical trials aimed at reducing chromosomal instability caused by aberrant centrosome behaviour, and exhibit incremental efficacy when combined with conventional chemotherapeutic agents, restoring therapeutic sensitivity and reducing tumour progression[287,443]. The FDA-approved Plk4 inhibitor CFI-400945 promotes defective centrosome duplication, cell death and inhibits tumour xenograft growth[447] and the FDA-approved Mps1/TTK inhibitor BAY1161909 (Empesertib)blocks spindle assembly, chromosome attachment and exhibits marked tumour suppressing activity when combined with paclitaxel[448]. FDA-approved Nutlins restores P53 function by inhibiting interaction with MDM2 and PRIMA-1, which targets Y220C mutant p53, restores wild-type p53 function reducing centrosome amplification[449,450]. Centrosome clustering, required for pseudo-bipolar spindle formation, leads to mitosis and aneuploid CSCs formation in centrosome-amplified polyploid giant cancer cells. This can be prevented by the FDA-approved small molecule PARP-6 inhibitor AZ0108, which promotes multipolar spindle formation and apoptosis in centrosome-amplified cells and exhibits anti-tumour effects in cancer models[451,452]. Centrosome clustering can also be disrupted by the investigational small molecule CCB02, which disrupts the interaction between centrosomal-P4.1-associated protein and b-tubulin, resulting in prolonged multipolar mitosis and death of centrosome-amplified cancer cells[453].
Alternative approaches
Targeting the tumour microenvironment:Reprogramming of the hypoxic tumour niche improves the efficacy of chemotherapy, fractionated radiotherapy and CAR Tcell immunotherapy[159,454], and represses hypoxia-induced CSCs phenotypes.Tumour oxygenation can be improved by hyperbaric oxygenation, intra-tumoural lipid-stabilized oxygen microbubble injection[455,456], nanoparticle-mediated reoxygenation, oxygen-generating methods[457] or artificial red cells[458]. The“normalization” of aberrant tumour vasculatures may also improve tumour oxygenation, reduce tumour progression and enhance therapeutic efficacy, stemming from observations that vascular destruction promotes tumour hypoxia, reduces therapeutic efficacy and facilitates metastatic progression. Tumour vessels are immature, permeable, tortuous, have aberrant basement membranes and lack cellular and matrix components required for maturation and function. This results in increased interstitial hypertension that promotes rapid drug-efflux and generates hypoxic niches, which promote and select polyploid giant cancer cell and CSC-like phenotypes.Vascular “normalization” requires delicate rebalancing of angiogenic factor/inhibitor equilibria by careful selection and dosage of antiangiogenic agents. Inhibition of VEGFA, VEGFRs or PHD2 proline hydroxylase inhibitors can prune immature permeable vessels, reduce permeability, improve vessel maturity, blood flow and oxygenation, decrease interstitial pressure to improve drug penetration and chemosensitivity, reduce metastatic progression and potentially reduce hypoxic nicheassociated CSC subpopulations[459,460]. Finally, tumour associated stromal and extracellular matrix barriers to CSC-targeted therapies delivered by nanoparticles or by engineered immune cells could be overcome using FAP+tumour fibroblasts targeted CAR T-cells, or other CAR cell types, engineered to express matrix degrading enzymes to disrupt tumour stroma and improve therapeutic access[159].
Engineered MSCs:The close relationship between MSCs and NB CSCs in tumourigenesis and metastatic progression[319-321] has also led to novel strategies that utilize MSCs to deliver anti-NB therapeutic agents. In the mouse TH-N-Myc NB model, IFNβtransduced mouse MSCs home to NBs, remain in tumours for up to 2 wk and augment tumour IFNβ levels[461]. Purified human adipose MSCs transfected with miR-124 and co-cultured with human M17 NB cells transfer miR-124 mimetic to NB cells by exosome delivery, resulting in miR-124-dependent reduced proliferation, increased apoptosis and neuronal differentiation, demonstrating that human adipose MSCs can be used effectively to deliver mimetic miRNAs to NB cells, supporting future therapeutic use of autonomous patient-derived MSCs as therapeutic vectors in NB[462]. Primary human MSCs engineered to express full length membrane-associated TRAIL[463], kill TRAIL receptor positive classic and primary NB cell lines, migrate to NB sites following intraperitoneal injection, and reduce NB xenograft growthin vivo,confirming drug delivery potential in NB[464]. However, the potential use of MSCs for drug delivery must take into account reports that MSCs also promote tumour growth and metastasis[465,466].
Oncolytic viruses:Genetically modified oncolytic virotherapy holds great promise as a novel anti-cancer therapy for eliminating CSCs[467]. The engineered oncolytic virus Nestin-targeted oHSV (rQNestin34.5) has been shown to kill doxorubicin-resistant CD133+SP CSC-like NB cells and to eradicate both bulk and tumour initiating tumour spheres, significantly delaying tumour formation inin vivomodels[468]. The first report of oncolytic virotherapy in NB was the direct intratumour injection of the Ad5/3-Cox2 oncolytic adenovirus, which induced a clinical response in a 6 year old stage IV NB patient[469]. The oncolytic adenovirus ICOVIR, that exhibits enhanced tumour selectivity by exploiting aberrant E2F expression, has been used to treat several children with stage IV refractory NB by intravenous injection of irradiationinactivated ICOVIR infected autologous MSCs, with complete 5 year remission in the only patient to exhibit a response[470]. In a recent first in-human and in-child trial study of autologous Icovir-5 infected MSC treatment of NB, two patients exhibited disease stabilization associated with low toxicity[471]. The engineered measles virus Edmonston strain has also been shown to have significant cytopathic effects on human primary and classical NB cell lines and xenograft models, inducing apoptotic cell death[472].
Nanoparticles for improving delivery and efficacy:Nanoparticle drug delivery facilitates better control of drug-release kinetics, improves bio-distribution and prolongs systemic and localized drug longevity. Mesoporous silica nanoparticles loaded with Notch signalling inhibitors or carrying γ-secretase inhibitors efficiently target tumour CSCs[473,474]. Salinomycin conjugated to a hyaluronic acid-based nanogel targets drug-resistant CD44+cells and enhances therapeutic efficacy[475].Nanoparticles coated with antibodies to CSC markers improve targeting, polymeric(D, L lactide-co-glycolide) nanoparticles coated with an anti-CD133 antibody and loaded with paclitaxel reduce tumourigenicity in mouse xenograft models[476], and Fe2O3nanoparticles coated with antibodies to ABCG2 transporter and loaded with paclitaxel target and kill myeloma cells[477]. With regard to NB, synthetic HDL nanoconjugates that target the SR-B1 receptor are taken up by validated NB CSCs, and reduce CSC viability, self-renewal, invasion and migrationin vitro,andin vivoexhibit tumour-uptake and reduce tumour growth,supporting further evaluation as a therapeutic NB CSCs inhibitor[478]. Mitochondrial-targeted silver nanoparticle cytotoxicity could be enhanced in NB cells by inhibitors of humanin expression,reported to protect NB cells against silver nanoparticle-induced toxicity[479]. Low nanoparticle penetration, due to low-level extravasation, has also been intelligently addressed, based upon greater understanding of the permeable and immature nature of tumour micro-vasculatures, with the development of gold nanoshell nanoparticles that seed tumour vasculatures for photothermal tumour ablation. This approach has demonstrated remarkable efficacy in mouse xenograft tumour models and in a clinical pilot study of low and intermediate risk prostate cancer, extolling a 94% success rate in tumour ablation[480].
CONCLUSION
The embryonic origin of NBs from NC stem/progenitor cells and their stemness gene expression patterns, indicate that NBs contain significant CSC-like subpopulations from the outset, which combined with properties of self-renewal, tumourigenicity,multipotency, therapy-resistance and reversible plasticity play central roles in NB heterogeneity, therapeutic-resistance, post-therapeutic-relapse, metastatic progression and unfavourable outcome. Conventional induction, consolidation and post-consolidation therapeutic strategies for high risk unfavourable NBs, induce initial clinical remission to states of no evidence or minimal residual disease, but also select and promote the formation of therapy-resistant polyploid giant cancer cells (PGCCs) and CSC subpopulations, adding to the probability of post-therapeutic relapse and metastatic progression. It is, therefore, of paramount importance that novel therapeutic strategies are developed to specifically target and eliminate PGCCs and CSC subpopulations in order to improve the current ≈ 8% 5-year overall survival rate in high risk NB[481].
As illustrated in this review, major advances in understanding the molecular protagonists, signalling pathways and mechanisms responsible for the induction,selection, maintenance and behaviour of NB PGCCs and CSCs have been made, and novel therapeutic strategies are currently being developed to target these populations.From our own perspective, observations that NF-κB-induced MMP-9 expression and MMP-9-dependent invasion associate with spontaneous S to I (CSC) phenotype conversion in NB cells[93,175], and the NB-associated oncogenic alternative TrkAIII splice variant promotes NB cell MMP-9 expression within a CSC context, links MMP-9 and stress-regulated alternative TrkAIII splicing to more aggressive NB CSC behaviour and identifies MMP-9 and TrkAIII as potential therapeutic NB CSC targets[72,73]. TrkAIII promotion of PI3K/Akt/NF-κB survival signalling, Bcl2 family expression[480], a survival-adapted ER-stress response[219], mitochondrial SOD2 expression[198] and pro-angiogenic MMP-9/VEGF/TSP-1 equilibrium, within a NB CSC context, furthermore, provide important insights into oncogene-regulated apoptosis evasion, survival within stressful tumour microenvironments, protection against mitochondrial ROS-induced death and angiogenesis, likely to extend to other splice-activated, mutated and fusion Trk oncogenes. Stress-regulated TrkAIIIdependent metabolic plasticity, unveils a mechanism for NB CSC metabolic adaption to changing conditions within tumour microenvironments[218], and TrkAIII promotion of centrosome amplification, unveils a mechanism for enhancing therapeutic resistance and promoting dormancy through NB PGCC formation[217],which upon escape from dormancy can generate blastomere-like CSCs with highly aggressive metastatic karyotypes. Furthermore, our recent discovery of a novel NFYAx splice variant, expressed by NBs and induced by doxorubicin, that selects NB CSC-like cells through differential cytotoxicity, unveils a potential mechanism for genotoxic drug-induced NB CSC selection and post-therapeutic relapse, which will be fully investigated in due course[136]. It is also our opinion that PGCCs formation and regulation of PGCC dormancy and escape from dormancy to generate potentially highly metastatic blastomere-stage CSCs, represent underestimated and understudied areas and should form the basis of novel pre-clinical models to study post-therapeutic NB relapse from PGCCs, with the aim of identifying single functional surface markers or marker-sets that uniformly and privately identify dormant NB PGCCs, counterparts that have escaped dormancy and their secondary CSC progeny, for better detection and therapeutic targeting and experimental therapeutics. Furthermore, current therapeutic strategies, which suffer from problems of delivery, uptake and efficacy due to aberrant tumour micro-vasculatures, physical barriers and acidic hypoxic microenvironmental conditions that promote drug-efflux, must also be improved to overcome these problems. In this review and within this context, we highlight the use of nanoparticles for improving drug kinetics or to facilitate thermal ablation; novel improved CSC-targeting CAR T-cells that modify inflammatory environments, modify tumour matrices and that can be switched off or eliminated to minimize side effects,with immune checkpoint inhibitors to improve infiltration; novel CAR NK, iNKT cells and engineered MSCs to target CSC niches; tumour oxygenation to reduce hypoxic CSC niches and enhance drug-sensitivity; centrosome dispersal and microtubule disrupting agents to perpetuate dormancy, and novel oncolytic viruses that exhibit improved CSC targeting and lytic activity (Figure 5). Continued research and development in these areas will hopefully lead to much needed improvements in event-free and overall survival in high risk unfavourable NBs.
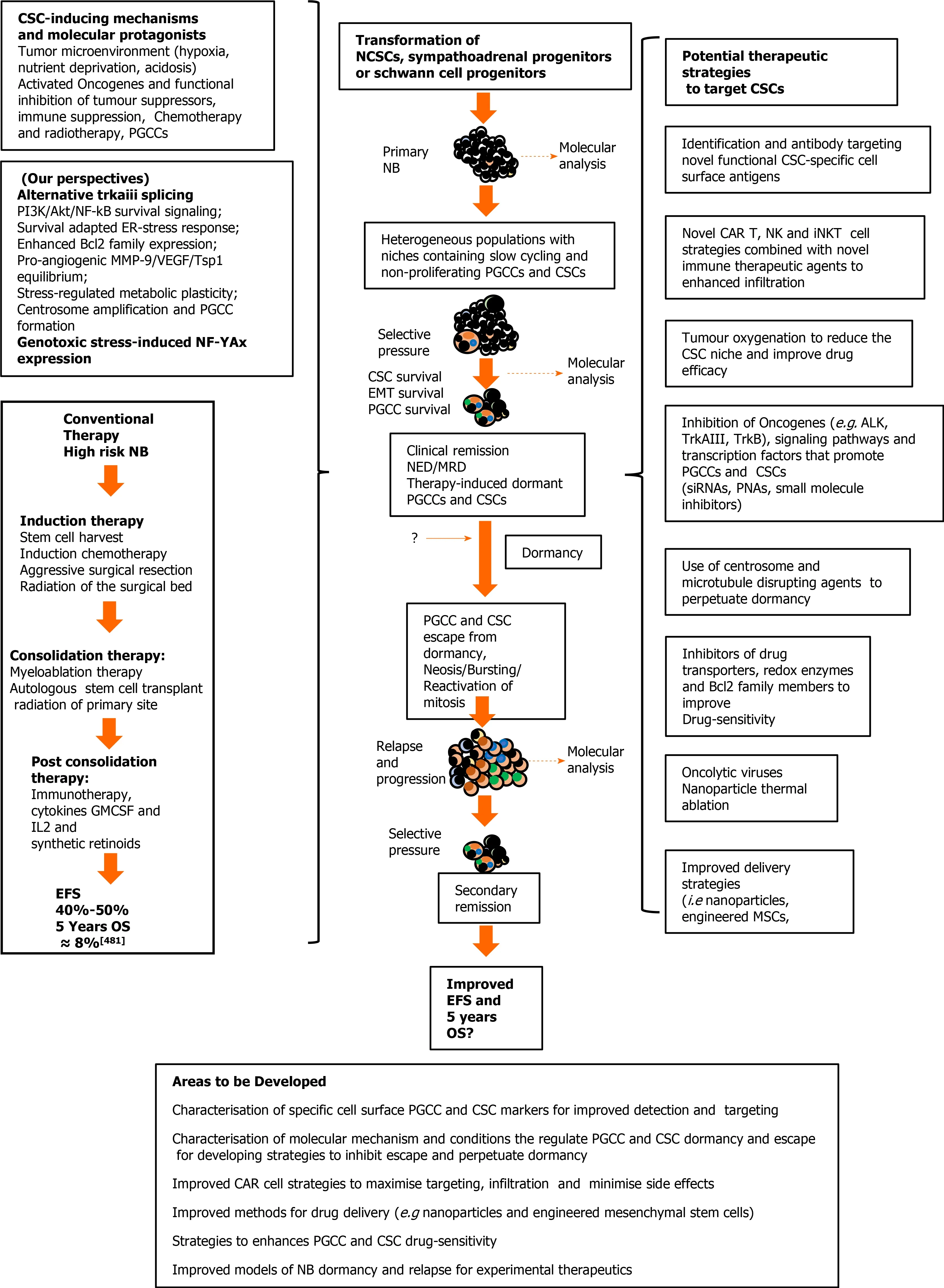
Figure 5 Cancer stem cell involvement in neuroblastoma relapse and progression, and potential therapeutic strategies. Neuroblastomas (NB)progression (center); mechanisms, molecular protagonists and conventional therapeutic strategies that select and induce the NB polyploid giant cancer cells (PGCC)and cancer stem cells (CSC), responsible for post-therapeutic relapse and low 5-year overall survival rates, in high risk unfavourable NB (right); potential therapeutic strategies to detect, target and eliminate NB PGCC and CSC subpopulations (left), and areas requiring further development (bottom). EFS: Event free survival; OS:Overall survival; CSC: Cancer stem cell; IL-2: Interleukin 2; GMCSF: Granulocyte Macrophage Colony-Stimulating Factor; NCSC: Neural crest stem cells; NB:Neuroblastomas; NK: Natural killer cells; ALK: Anaplastic lymphoma kinase; siRNA: Small interfering ribonucleic acid; PNA: Peptide nucleic acid; MSC: Mesenchymal stem cells; PGCCs: Polyploid giant cancer cells.
杂志排行

World Journal of Stem Cells的其它文章
- Epigenetic modulators for brain cancer stem cells: Implications for anticancer treatment
- Roles of mitochondrial unfolded protein response in mammalian stem cells
- Stem cell therapies in tendon-bone healing
- Exosomal microRNAs from mesenchymal stem/stromal cells:Biology and applications in neuroprotection
- Immunotherapy against programmed death-1/programmed death ligand 1 in hepatocellular carcinoma: Importance of molecular variations, cellular heterogeneity, and cancer stem cells
- Bone marrow mononuclear cells for joint therapy: The role of macrophages in inflammation resolution and tissue repair