Epigenetic modulators for brain cancer stem cells: Implications for anticancer treatment
2021-07-30LuanaAbballeEvelinaMiele
Luana Abballe, Evelina Miele
Luana Abballe, Evelina Miele, Department of Pediatric Hematology/Oncology and Cellular and Gene Therapy, Bambino Gesù Children's Hospital, IRCCS, Rome 00165, Italy
Abstract Primary malignant brain tumors are a major cause of morbidity and mortality in both adults and children, with a dismal prognosis despite multimodal therapeutic approaches. In the last years, a specific subpopulation of cells within the tumor bulk, named cancer stem cells (CSCs) or tumor-initiating cells, have been identified in brain tumors as responsible for cancer growth and disease progression. Stemness features of tumor cells strongly affect treatment response, leading to the escape from conventional therapeutic approaches and subsequently causing tumor relapse. Recent research efforts have focused at identifying new therapeutic strategies capable of specifically targeting CSCs in cancers by taking into consideration their complex nature. Aberrant epigenetic machinery plays a key role in the genesis and progression of brain tumors as well as inducing CSC reprogramming and preserving CSC characteristics. Thus, reverting the cancer epigenome can be considered a promising therapeutic strategy. Three main epigenetic mechanisms have been described: DNA methylation, histone modifications, and non-coding RNA, particularly microRNAs. Each of these mechanisms has been proven to be targetable by chemical compounds, known as epigeneticbased drugs or epidrugs, that specifically target epigenetic marks. We review here recent advances in the study of epigenetic modulators promoting and sustaining brain tumor stem-like cells. We focus on their potential role in cancer therapy.
Key Words: Cancer stem cells; Epigenetics; Brain tumors; Epigenetic drugs; Histone deacetylase inhibitors; DNA methyltransferase inhibitors
INTRODUCTION
Primary malignant brain tumors are a heterogeneous group of tumors arising from the brain parenchyma and its surrounding structures. They represent a major cause of morbidity and mortality in both adults and children. In particular, brain tumors are the most frequently reported solid malignancies in children, accounting for up to 20%of childhood cancers[1]. Brain cancers require a multimodal treatment that includes surgical resection, chemotherapy, and radiation therapy[2]. This therapeutic strategy includes a particularly invasive surgery as well as brain irradiation and can lead to long-lasting side effects, significantly decreasing the patient’s quality of life.
Brain tumors can harbor genetic and epigenetic alterations that make them resistant to conventional pharmacological treatment[3]. Current therapeutic approaches do not consider the presence of a specific subpopulation of cells within the tumor bulk,known as cancer stem cells (CSCs) or tumor-initiating cells. These cells are not responsive to conventional treatment, promoting tumor relapse[4]. In this scenario, it is of fundamental importance to seek alternative therapeutic strategies that take into account the genetic and epigenetic alterations of the tumor as well as the presence of tumor-initiating cells. Nowadays, to address this challenge, the epigenetic profile of CSCs is being thoroughly studied and several epigenetics drugs are being tested inin vitroandin vivostudies on CSC cultures[5].
This review aims to summarize and organize the current epigenetics strategies for eradicating brain CSCs based on CSC epigenetic modulators.
Brain cancer stem cells
CSCs are considered the “reservoirs” of the tumor. They are defined as a small subset of stem cells capable of proliferating and generating diverse and heterogeneous cell types that constitute the whole tumor[6]. CSCs reside in specific anatomical areas called “niches”, where they interact with the microenvironment surrounding them[7].Like their normal tissue counterpart, CSCs exhibit “stem-like” characteristics: (1) Cell quiescence: A way to preserve self-renewal and to avoid genetic perturbation that could occur during cell division; (2) Self-renewal capacity: The ability to proliferate symmetrically and asymmetrically; (3) Multipotency: The ability to give rise to heterogeneous cells with different proliferative potential; (4) Migration: The ability to migrate and disseminate; (5) Tissue regeneration: The ability to give rise to new tumoral tissues; and (6) Communication: The ability to interact with the microenvironment.
The substantial difference between CSCs and normal tissue stem cells is that proliferation/death signals are aberrant and dysregulated in cancer, whereas normal tissue stem cells can maintain physiological homeostasis.
CSCs were discovered for the first time in acute myeloid leukemia[8]. Subsequently,this tumor-initiation subpopulation was also identified in many solid tumors,including brain cancers[9,10]. CSCs have been discovered and isolated in major brain tumors [e.g., medulloblastoma (MB), gliomas, ependymoma][11-13] from both pediatric and adult patients[14-16].
In the first instance, cell surface markers were used to identify this subset of tumorinitiating cells. The cell surface antigen CD133 has been most frequently used to mark CSCs in various solid tumors[17]. In the context of the brain, Singhet al[11]demonstrated that CD133+human brain tumor cells were able to initiate and recapitulate the original tumor inin vivomouse models. Further study demonstrated that a high expression level of CD133 was correlated with poor prognosis in brain cancer patients, reinforcing CD133’s role as a “brain stemness” marker[18].
The accuracy of this detection method remains very controversial because cell surface markers evolve rapidly in response to different environmental stimuli, disease states, and tumor progression[19,20]. Moreover, brain CSCs and neural stem cells share common phenotypic markers (e.g., CD133, CD15, CD44)[21]. Hence, the markerdependent identification method alone is insufficient to discriminate correctly CSCs from normal tissue ones. Recent studies[22-24] investigate complementary methods that consider cell dysregulated survival pathways and genetic and epigenetic signatures. As already described by Abbaszadeganet al[23], the gold standard strategy to identify efficiently brain CSCs is to test theirin vivotumorigenicity. Limiting dilution assay is the best tumorigenicity method that is commonly used for evaluation of CSCs frequency. However, this method presents some critical points, being influenced by the number of the cells, the implantation site, and growing time of incubation. Moreover, it is not feasible on large scale studies. Complementaryin vitrofunctional assays could be used to identify CSCs based on: (1) Their intrinsic properties (e.g., self-renewal, asymmetrical division, slow proliferation phenotype, and aldehyde dehydrogenase 1 expression); and (2) Their survival pathways (e.g., Wnt/βcatenin, Hedgehog, and Notch signaling pathway), in terms of expression of transcription factors/key proteins/microRNAs (miRNAs). Among the recently developed approaches to isolate CSCs, there are next generation sequencing (NGS)technologies. For example, Jonassonet al[24] isolated the stem-like subpopulation using a functional cellular assay that enriches for cells that can self-renew and differentiate, combined with NGS technologies (single-cell RNA sequencing) to identify CSCs[22-24]. Moreover, Rodriguez-Meiraet al[25] in their scientific work developed an NGS platform that combines single-cell RNA-seq with mutational analysis allowing the identification of distinct subclones of cancer cells[26]. This evidence suggests that a combination of cell surface markers and functional assays provide an efficient tool for their identification.
CSCs’ properties have profound implications for treatment response due to their ability to evade conventional therapy and cause subsequent tumor relapse[27,28]. In this case, it is crucial to identify new treatment strategies that take into consideration the complexities of CSCs. Currently, global researchers are making great efforts to understand the biological properties of CSCs and develop new therapeutic approaches targeting CSCs.
EPIGENETIC MODULATORS
The term "epigenetics" is used to refer to information that controls gene expression that is stable and inheritable during cell division and happens without changes in the DNA sequence. Aberrant epigenetic landscapes control cell fate specification,promoting tumor initiation and progression[29,30].
Epigenetics controls gene expression through three main mechanisms: DNA methylation, histone modifications, and non-coding RNA, particularly miRNAs[31](Figure 1).
DNA methylation is the most studied epigenetic modification. It is associated with transcriptional inactivation and closed chromatin structure, mainly regulating gene silencing or repression. It depends on the action of specialized enzymes, known as DNA methyltransferases (DNMTs) that transfer a methyl group from S-adenosyl methionine to the fifth carbon of the pyrimidine ring of a DNA base cytosine. DNA methylation occurs mainly at CpG dinucleotides, which are concentrated in specific regions of DNA called “CpG islands”. CpG islands are located at gene promoters, in regulatory regions, and in gene bodies[32], but DNA methylation could also be present in non-coding DNA sequences such as repetitive elements, transposons, non-coding RNAs, and introns[33,34].
A methylation pattern represents the fingerprint of a cell. It is established during early embryogenesis, and it is maintained during cell division by the action of DNMTs.
Methylation status is altered in cancers, mainly through two mechanisms: Regional hypermethylation and global hypomethylation[35]. DNA hypermethylation involves the CpG islands, which are usually unmethylated in normal cells. The result of DNA hypermethylation is the transcriptional repression of tumor-suppressor and tissuespecific genes, and the inactivation of miRNA, which are involved in the initiation and progression of cancer[34]. This mechanism occurs in different stages of carcinogenesis including CSC formation[36]. Conversely, global hypomethylation consists of the loss of the methyl group on cytosine, mainly in repetitive elements across DNA.Hypomethylation was one of the first epigenetic features discovered in human cancers[37], causing the reactivation of methylated regions of DNA such as transposons,introns, and germ-line genes that are silent in differentiated cells[34,38]. In high-grade cancers, such as glioblastoma (GBM), hypomethylation is a mechanism used by CSCs to reactivate key stem cell genes[39].
Abnormalities of DNA methylation are early events in pre-malignant transformation and are maintained in the global tumor population. However, the epigenome is in continuous evolution, and some of the changes are detectable in later steps of tumorigenesis as a result of positive selection. In this way, epigenome contributes to tumor heterogeneity and plasticity, which gives rise to a heterogeneous tumor composed of different cell subpopulations, one of them could have “stem-like” features.Additionally, compared to the bulk tumor, CSCs could acquire further epigenetic alterations in response to stress of different nature (e.g., chemotherapy/radiotherapy,chronic inflammation, and environmental exposures), contributing to tumor relapse[40].
The development of powerful “next-generation” techniques makes it possible to describe the entire epigenome of cells or tissues[38]. Capperet al[41] developed a DNA methylation-based classification for central nervous system cancers that allows discrimination between different subtypes of tumors, some of them previously considered as homogeneous diseases.
Histones are subject to reversible post-translational modifications (PTMs) that cooperate to govern the chromatin state. Histone PTMs influence chromatin structure that is conducive to the expression or repression of target genes. Histone aminoterminal regions can undergo diverse PTMs: Methylation, acetylation, phosphorylation, ubiquitylation, biotinylation, sumoylation, and ADP-ribosylation[42] that work in concert to define the chromatin status of the specific region of DNA.
Histone PTMs are controlled by different enzymes: "Writers" catalyze histone modifications; “erasers” cut histone modifications; and “readers” translate the PTMs’language into cellular signals. All enzymes work together in a very specific manner to create the “histone code”.
Histone methylation and acetylation are the best-known PTMs on histone residues.Histone methylation is a reversible mechanism that occurs on lysine and arginine residues, leading to a different degree of methylation. Up to three methyl groups can be added to a single lysine residue (un-, mono-, di-, and tri-methylated states) and up to two groups to a single arginine residue (mono- and di-methylated states).Methylation can be either an activating or a repressive mark, depending on the target histone and lysine residue: For example, histone H3 Lysine 27 (H3K27) and histone H4 Lysine 20 (H4K20) are usually associated with gene silencing, while H3 Lysine 4(H3K4) and H3 Lysine 36 (H3K36) are transcriptional activation marks. Histone acetylation is associated with relaxed chromatin structure and promotes the binding of transcription factors and RNA polymerase to DNA, resulting in the activation of gene expression[42].
Recent efforts have tried to highlight the role of epigenetic modulators in the development and plasticity of brain tumor CSCs[43,44]. In GBM CSCs (GSCs), Liauet al[45] showed in patient-derived GSCs, which effectively initiate tumors in mice models, that KDM6-mediated demethylation of histone H3 Lysine 27 trimethylation(H3K27me3), a repressive mark, has a central role in the maintenance and persistence of GSCs. Maramponet al[46] demonstrated the fundamental role of two histone deacetylases (HDAC4 and HDAC6) in regulating the DNA repair machinery, survival,and stemness characteristics of radioresistant GSCs derived from U87MG and U251MG human GMB cell lines subjected to irradiation. Moreover, a recent study by Banelliet al[47] on human GSCs derived from human tumors describes a subset of GSCs resistant to chemotherapy that are particularly dependent on KDM5A action,making them strongly sensitive to HDAC inhibitor (HDACi) treatments in term of cell viability, percentage of apoptotic cells, and reducing capacity of clonal growth. In medulloblastoma, pharmacological inhibition of histone methyltransferase enhancer of zeste 2 (EZH2) impairs proliferation and self-renewal of human and mouse stem-like cells, derived from primary human Sonic Hedgehog MB (SHH-MBs) and from tumors arisen in Ptc+/- mice, inin vitroandin vivostudies[48].
miRNAs and epigenetic modulators create a miRNA-epigenetic feedback loop: On the one hand, miRNA can regulate the transcription of epigenetics-associated enzymes, while on the other miRNA transcription is under the control of epigenetics machinery (DNA methylation, histone PTMs, and RNA modifications)[49].
In literature, there are several examples of miRNA-epigenetic machinery in CSCs.For example, Ferrettiet al[50] described the role of the miR-326-ARRB1-E2F1 axis in medulloblastoma CSCs survival. For this study, MB CSCs were derived from tissues freshly resected from pediatric patients and cultured in CSC-enriched cultures.Notably, miR-326 is already described as onco-suppressor miRNA[50]. It is involved in neuronal differentiation, and its expression is under control of the histone-lysine Nmethyltransferase EZH2. High levels of EZH2 are responsible for the presence of H3K27me3 on the promoter region of miR-326[22]. In another study, Lizarte Netoet al[51] investigated the role of miR-181d and the methylation status of the MGMT gene in response to pharmacological treatment in CD133+ GBM CSCs. Their results showed an increase of miR-181d and MGMT transcription levels after treatment, due to cellular mechanism of drug resistance[51].
The tumor microenvironment (TME) also acts as an epigenetic regulator for cancer cells. TME communicates with cancer cells through extracellular vesicles secreted by many TME cell types that contain various mediators including proteins and nucleic acids. Also, miRNAs can be charged in the extracellular vesicles and thereby alter the epigenome of the recipient cancer cell[52].
All this evidence makes brain CSCs an excellent candidate for pharmacological epigenetic treatments.
TRANSLATIONAL SIGNIFICANCE OF EPIGENETICS: EPIDRUGS
Epigenetics-based drugs (epidrugs) are chemical compounds that specifically target epigenetic marks (Figure 1). CSCs could acquire mutations in epigenetic marks or changing in methylation/acetylation status, or again in miRNAs signature, that make them sensitive to epigenetics-based drugs’ approaches. Epidrugs are based on the idea that by changing the epigenome of CSCs, it may be possible to remodel the fate of cells from CSCs to differentiated tumor cells[53-55]. Epidrugs are used as monotherapy or in combination with chemo-/radio-therapy to target both CSCs and bulk tumors.Today, several HDAC and DNMT inhibitors are in different clinical trial phases.
HDACi
Among the various epigenetic modulators, the acetylation state of histone proteins is one of the major targets for anticancer therapy. Histone acetylation is controlled by two types of enzymes: HDACs and histone acetyltransferases (Figure 1).
Human HDACs are categorized into four different classes: Class I (HDAC1, 2, 3,and 8), class IIa (HDAC4, 5, 7, and 9), class IIb (HDAC6 and 10), class III (sirt 1-7), and class IV (HDAC11). HDACs are enzymes that remove the acetyl group from lysine residues on histones, which compact the chromatin structure into a non-permissive state, resulting in transcriptional repression.
Based on isoform selectivity, HDACi can be classified as: (1) Pan-inhibitors, if they act against all HDACs; or (2) HDAC isoform-selective inhibitors if they target a specific HDAC class[56]. Pan-inhibitors are in turn classified according to their chemical structure as: (1) Hydroxamic acids; (2) Aliphatic carboxylic acids; (3)Benzamides; (4) Cyclic peptides; or (5) Sirtuin inhibitors.
HDACs control several cellular mechanisms. They have been implicated in different types of cancers[46], and several HDACs are overexpressed in brain cancers[57,58]. For example, Staberget al[59] found an up-expression of HDAC1, 3, and 6 in 21 primary GBM cell cultures, grown as neurospheres, compared to non-neoplastic brain control cells (normal human astrocytes), and confirmed these findings in a panel of primary GBM tissue samples compared to normal brain tissues. They demonstrated the efficacy of HDACi therapy (trichostatin A) in GBM treatment inin vitroexperiments.
HDACi exhibit anti-cancer activity against CSCs in tumors with a predominant stem-like population such as brain cancers. HDACi target the escape mechanism of CSCs, reversing chemo-radio-therapy resistance by inducing cell differentiation,apoptosis, inhibition of angiogenesis, and upregulation of tumor suppressor genes[60].
In line with this observation, da Cunha Jaegeret al[61] demonstrated that HDACi and mitogen-activated protein kinase/extracellular signal-regulated kinase inhibitors could modulate the stemness markers (BMI1 and CD133), viability, and neurosphere formation capacity of MB cell lines (DAOY and D283 cultured in serum-free sphereinduction medium). Their results proposed HDACi as a valid candidate for MB CSC treatment[61]. Coniet al[62] reported antitumor effects of selective HDAC1 and HDAC2 inhibition on SHH-MB cells and mouse models. Also, in diffuse intrinsic pontine glioma tumor models (patient-derived neurospheres, xenografts, and allografts), combination of HDAC and AXL inhibition modulate the H3K27M epigenetic mark resulting in a down-regulation of stemness markers (SOX2 and its target genes directly correlated with stem cell characteristics)[63].
Several HDACi are emerging as promising anticancer drugs in clinical trials for brain cancer treatment, both as combinatorial- and as mono-therapy[64]. Table 1 shows the HDACi used in clinical trials for brain cancers (Vorinostat, Valproic acid,Panobinostat, and Entinostat). Most clinical trials are focused on the usage of Vorinostat (suberoylanilide hydroxamic acid or SAHA), a potent inhibitor of HDAC classes I and II. Vorinostat was initially approved by the Food and Drug Administration for treating refractory cutaneous T-cell lymphoma, and its use was subsequently extended to various solid cancers[56]. In a phase 2 clinical trial,Vorinostat was tested as a mono-therapeutic agent in GBM treatment to target both CSCs and differentiated cancer cells. In fact, preclinical studies demonstrated the efficacy of Vorinostat in reducing EZH2 and CD133 stemness marker in patientderived GBM CSCs[65] and in modulating senescenceviathe p38-p53 axis and inducing apoptosis in GBM cell lines cultured in serum-free sphere-induction medium[66]. Sunget al[67] demonstrated that a combination of Vorinostat with melatonin can overcome the pharmacological resistance of human GBM CSCs, reducing self-renewal and proliferation of stem cell compartments and increasing apoptosis markers(cleaved poly-ADP ribose polymerase and p-γH2AX).
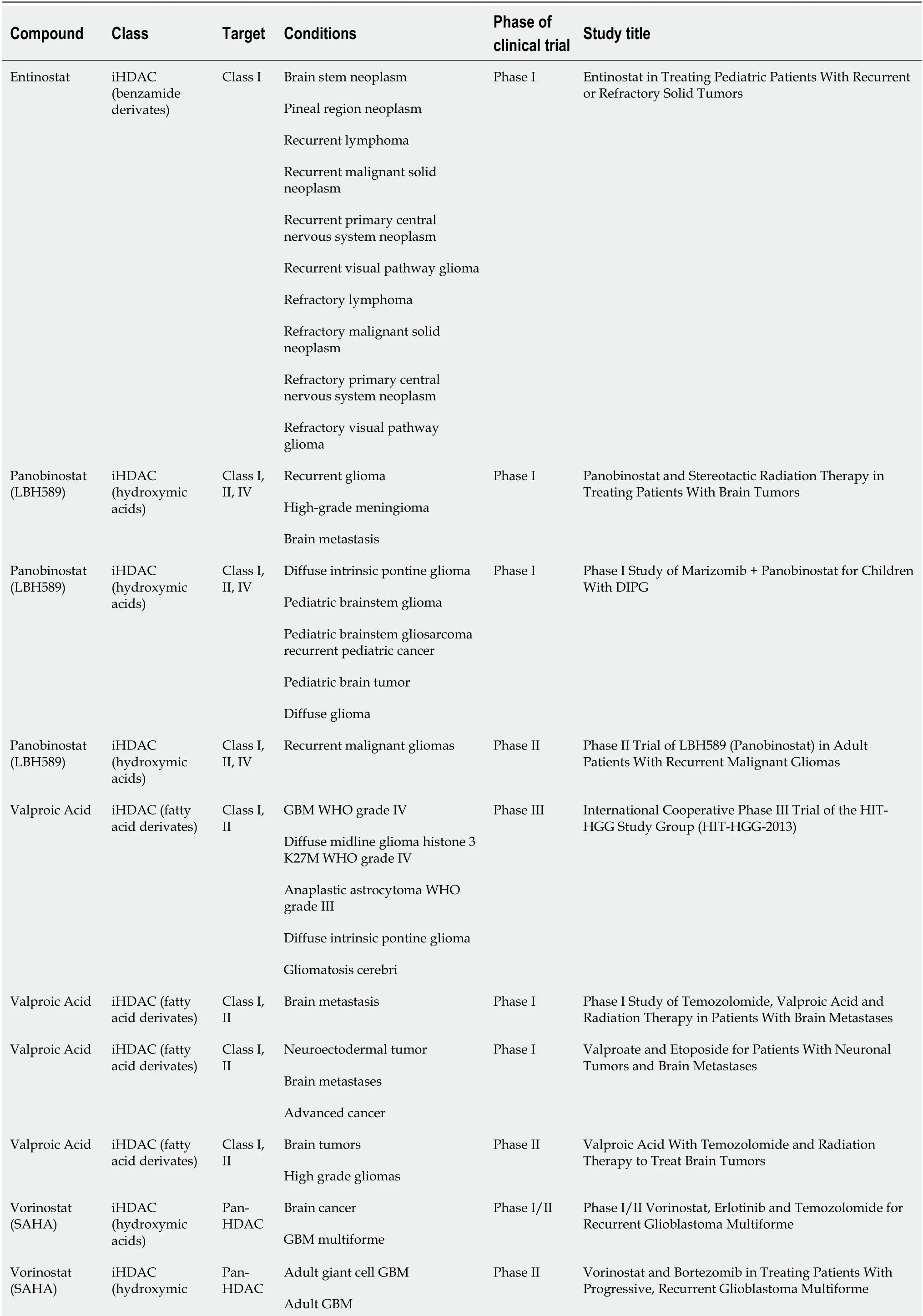
Table 1 Histone deacetylases inhibitors in clinical trial for brain cancer treatment
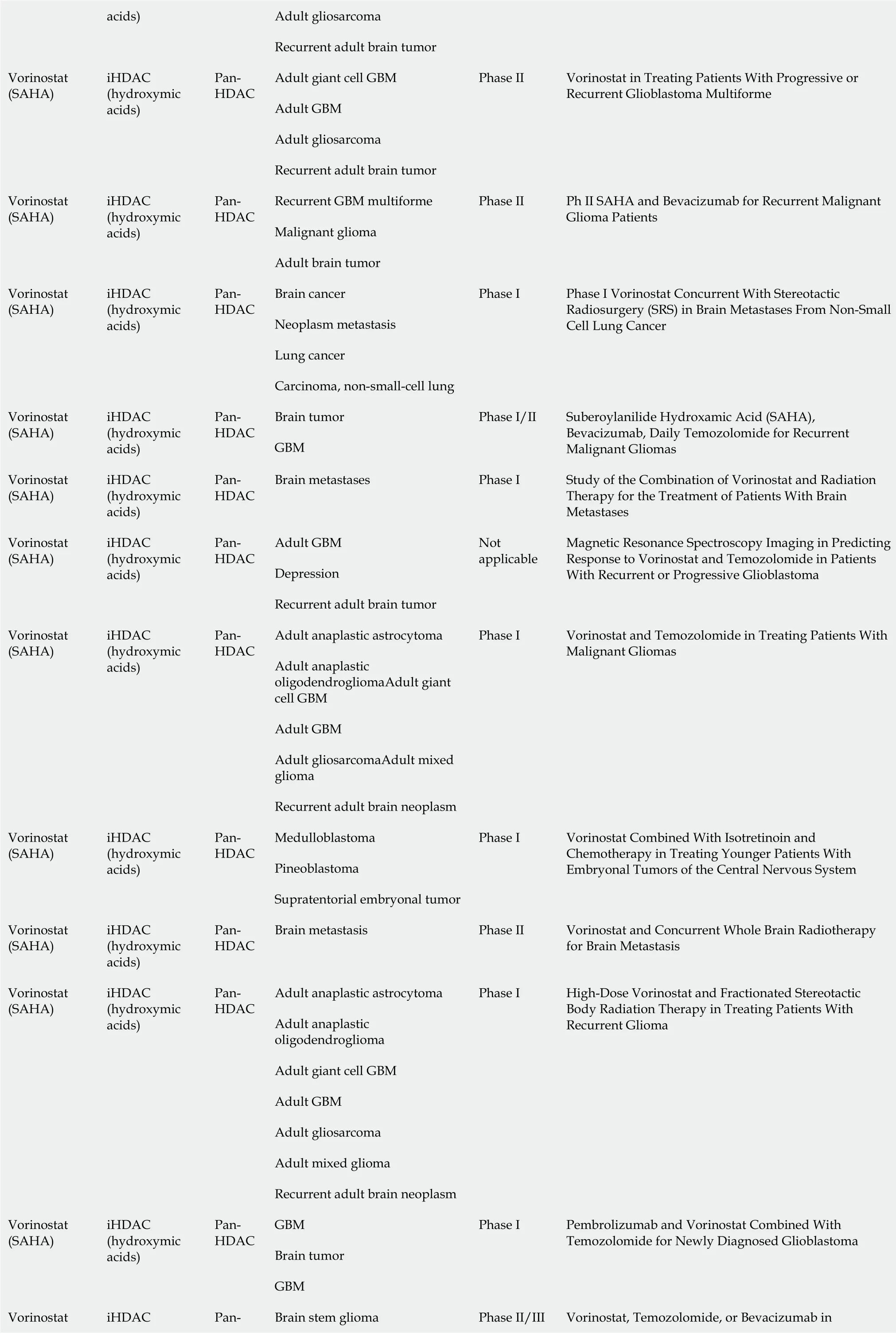
Adult gliosarcoma acids)Recurrent adult brain tumor Adult giant cell GBM Adult GBM Adult gliosarcoma Vorinostat(SAHA)iHDAC(hydroxymic acids)Pan-HDAC Recurrent adult brain tumor Phase II Vorinostat in Treating Patients With Progressive or Recurrent Glioblastoma Multiforme Recurrent GBM multiforme Malignant glioma Vorinostat(SAHA)iHDAC(hydroxymic acids)Pan-HDAC Adult brain tumor Phase II Ph II SAHA and Bevacizumab for Recurrent Malignant Glioma Patients Brain cancer Neoplasm metastasis Lung cancer Vorinostat(SAHA)iHDAC(hydroxymic acids)Pan-HDAC Carcinoma, non-small-cell lung Phase I Phase I Vorinostat Concurrent With Stereotactic Radiosurgery (SRS) in Brain Metastases From Non-Small Cell Lung Cancer Brain tumor Vorinostat(SAHA)iHDAC(hydroxymic acids)Pan-HDAC GBM Phase I/II Suberoylanilide Hydroxamic Acid (SAHA),Bevacizumab, Daily Temozolomide for Recurrent Malignant Gliomas Vorinostat(SAHA)iHDAC(hydroxymic acids)Pan-HDAC Brain metastases Phase I Study of the Combination of Vorinostat and Radiation Therapy for the Treatment of Patients With Brain Metastases Adult GBM Depression Vorinostat(SAHA)iHDAC(hydroxymic acids)Pan-HDAC Recurrent adult brain tumor Not applicable Magnetic Resonance Spectroscopy Imaging in Predicting Response to Vorinostat and Temozolomide in Patients With Recurrent or Progressive Glioblastoma Adult anaplastic astrocytoma Adult anaplastic oligodendrogliomaAdult giant cell GBM Adult GBM Adult gliosarcomaAdult mixed glioma Vorinostat(SAHA)iHDAC(hydroxymic acids)Pan-HDAC Recurrent adult brain neoplasm Phase I Vorinostat and Temozolomide in Treating Patients With Malignant Gliomas Medulloblastoma Pineoblastoma Vorinostat(SAHA)iHDAC(hydroxymic acids)Pan-HDAC Supratentorial embryonal tumor Phase I Vorinostat Combined With Isotretinoin and Chemotherapy in Treating Younger Patients With Embryonal Tumors of the Central Nervous System Vorinostat(SAHA)iHDAC(hydroxymic acids)Pan-HDAC Brain metastasis Phase II Vorinostat and Concurrent Whole Brain Radiotherapy for Brain Metastasis Adult anaplastic astrocytoma Adult anaplastic oligodendroglioma Adult giant cell GBM Adult GBM Adult gliosarcoma Adult mixed glioma Vorinostat(SAHA)iHDAC(hydroxymic acids)Pan-HDAC Recurrent adult brain neoplasm Phase I High-Dose Vorinostat and Fractionated Stereotactic Body Radiation Therapy in Treating Patients With Recurrent Glioma GBM Brain tumor Vorinostat(SAHA)iHDAC(hydroxymic acids)Pan-HDAC GBM Phase I Pembrolizumab and Vorinostat Combined With Temozolomide for Newly Diagnosed Glioblastoma Brain stem glioma Vorinostat iHDAC Pan-Vorinostat, Temozolomide, or Bevacizumab in Phase II/III
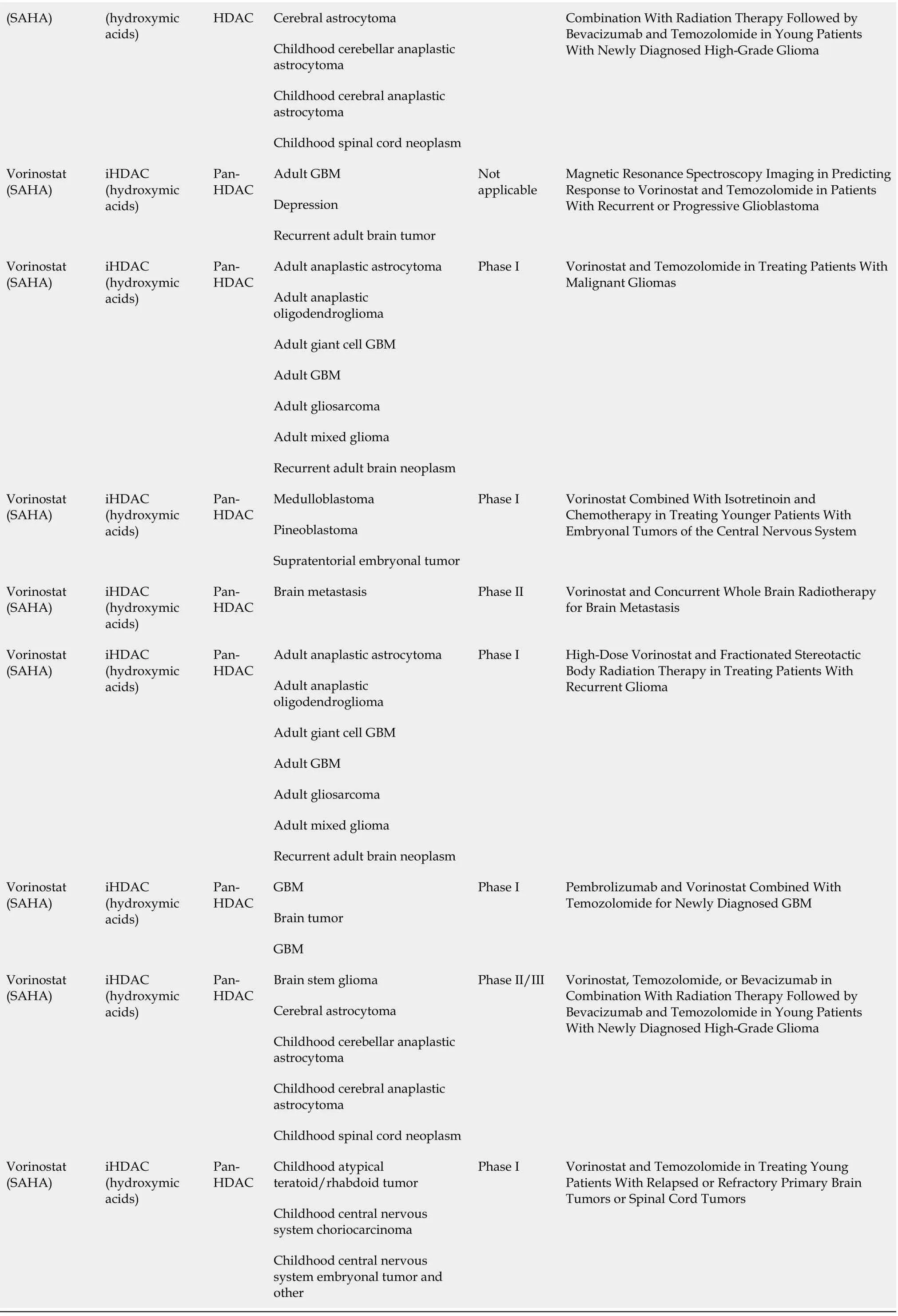
HDAC: Histone deacetylases; iHDAC: HDAC inhibitors; DIPG: Diffuse intrinsic pontine gliomas; WHO: World Health Organization; Ph: Phase; SRS:Stereotactic radiosurgery; GBM: Glioblastoma.
Vorinostat acts by inducing the acetylation of proteins, including histones and transcription factors at both transcriptional and non-transcriptional levels, leading to different cellular effects[68]. Vorinostat, like other pan-HDACi, has variable activity against HDAC isoenzymes, some of which are important in antitumor response.
Vorinostat shows a different toxicity profile compared to classical chemotherapeutic drugs. In fact, common pan-HDACi side effects include fatigue, nausea/vomiting,anemia, anorexia, increased blood urea, hyperglycemia, and thrombocytopenia. Some of these adverse effects can be routinely managed by physicians, but some others need more careful monitoring[69]. However, side effects are dose-dependent and Vorinostat is effective at very low concentrations[70]. There are several reasons behind the epidrugs’ toxicity. It is partly due to “on-target” effects, which could be explained with the concept of “pleiotropy”. Specifically, a single target gene could be involved in different signaling and controls multiple phenotypic effects. Another reason is “off-target” effects. Epi-drugs are designed to inhibit aberrant epigenetic enzymes, but it is known that they could also affect other classes of substrates belonging to unintended cellular pathways, at both intracellular and extracellular levels[71].
In medulloblastoma, the administration of epigenetic modifiers such as Vorinostat and Valproic acid shows radiosynergistic action on the proliferation and cloning capacity of three human MB cell lines (DAOY, MEB-Med8a, and D283-Med), offering a new opportunity to treat MB patients[72].
DNA methyltransferase inhibitors
The readout of DNMTs is hypermethylation of the DNA sequence and resulting silencing of gene expression. Aberrant DNA methylation has been associated with different stages of cancers. This evidence supports a rationale for using DNMT inhibitors (DNMTi) in cancer treatment (Figure 1).
There are two different classes of DNMTi: (1) Nucleoside analogues, which could be considered analogues to cytosine and act as a natural substrate for DNMT (e.g., 5-azacytidine); and (2) Nonnucleoside compounds, which inhibit DNA methyltransferase activity through mechanisms other than DNA incorporation.
This second class of DNMTi appears to be less toxic and more stable than nucleoside analogues[5,73,74], and Valente and colleagues have focused on the study of nonnucleoside compounds[75].
The archetypal DNA methyltransferase inhibitor is 5-azacytidine (also known as 5-aza), it is a nucleoside inhibitor that is incorporated into the DNA sequence and covalently bonds to and inactivates DNA methyltransferase. As shown in Table 2, 5-azacytidine has been tested in different phase I trials against various brain cancers,particularly in recurrent brain tumors, GBM, and ependymoma.
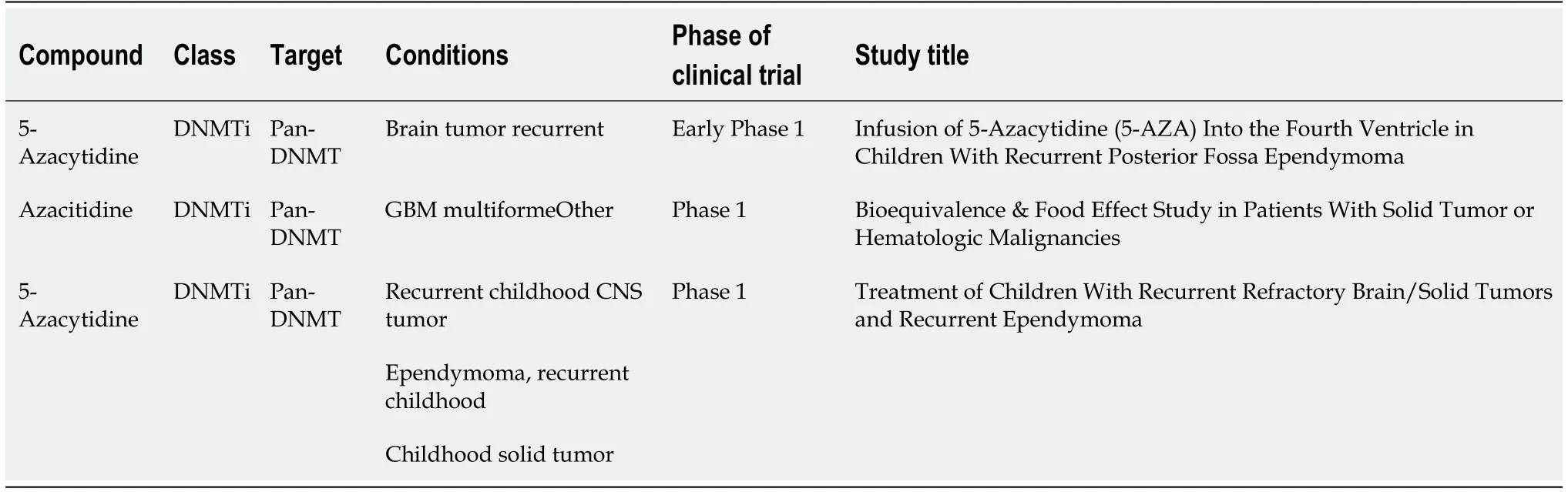
Table 2 DNA methyltransferase inhibitors in clinical trial for brain cancer treatment
Several studies have also demonstrated the differentiation potential of DNMTi in cancer therapy. Liaoet al[76] showed that decitabine, an analogue of cytosine, in combinatorial treatment with a differentiation drug, could provide an effective route to enhancing cell differentiation (oligodendrocyte-like morphology and mRNA expression of terminal differentiation marker MBP) and inhibiting cell growth in two malignant glioma human cell lines. Andradeet al[77] tested the efficacy of zebularine,another DNMTi, in four pediatric SHH-MB cell lines (DAOY, ONS-76, UW402, and UW473). Zebularine decreased MB cell growth by targeting the Sonic Hedgehog pathway’s components (GLI1, SMO and PTCH1), evaluated at transcriptional levels.This provides a rationale for furtherin vivoinvestigation into the combination of zebularine with chemotherapy[77]. Valenteet al[75] tested two nonnucleosides DNMTi(compounds 2 and 5) in mouse MB stem cells isolated from fresh tumor specimens from Ptch1+/- mouse models; compound 2 significantly blocked cell proliferation,while compound 5 was stronger in differentiation potential evaluated by both βIIItubulin reverse transcriptase polymerase chain reaction and morphology images.
CONCLUSION
Aberrant epigenetic regulation has emerged as a key player in the genesis and progression of brain tumors, influencing malignant phenotypes at various stages of the disease and possibly underlying individual variability in drug response. In particular,the epigenome of brain tumors can be modulated by both cell-intrinsic (e.g., mutations)and cell-extrinsic (e.g., microenvironment) mechanisms, favoring those characteristics of CSCs responsible for cancer growth and disease progression. Reprogramming the epigenetic landscape in the cancer epigenome is among the most promising target therapies, both as a treatment itself and for reversing drug resistance. In this review,we discussed how epigenetic alterations regulate the "stem-like" properties of CSCs and the epigenetic drugs available to blockade epi-mutations. We have reported several examples of epidrugs in Phase I/II clinical trials, providing evidence on the benefit of using epidrugs as single agents or in combination-therapy in brain tumors.We believe this is thus a viable avenue for clinical trials aiming at the development of more affordable and efficient anticancer drugs and treatments.
ACKNOWLEDGEMENTS
We thank Ralf Mouthaan for manuscript editing.
杂志排行

World Journal of Stem Cells的其它文章
- Mechanisms involved in selecting and maintaining neuroblastoma cancer stem cell populations, and perspectives for therapeutic targeting
- Roles of mitochondrial unfolded protein response in mammalian stem cells
- Stem cell therapies in tendon-bone healing
- Exosomal microRNAs from mesenchymal stem/stromal cells:Biology and applications in neuroprotection
- Immunotherapy against programmed death-1/programmed death ligand 1 in hepatocellular carcinoma: Importance of molecular variations, cellular heterogeneity, and cancer stem cells
- Bone marrow mononuclear cells for joint therapy: The role of macrophages in inflammation resolution and tissue repair