ACE2 downregulation promotes thrombosis and cardiac injury in COVID-19 patients
2021-07-20DhanasekaranSivaramanKagithakaraVajraveluLeelaVenkatesaluVenugopal
Dhanasekaran Sivaraman, Kagithakara Vajravelu Leela, Venkatesalu Venugopal
1Department of Pharmacology and Toxicology, Centre for Laboratory Animal Technology and Research, Sathyabama Institute of Science and Technology, Jeppiaar Nagar, Chennai, Tamil Nadu 600119, India
2School of Pharmacy, Sathyabama Institute of Science and Technology, Jeppiaar Nagar, Chennai, Tamil Nadu 600119, India
3Chinese Association for Laboratory Animal Sciences, Chaoyang District, Beijing 100021, China
4Department of Microbiology, SRM Medical College Hospital and Research Centre, Tamil Nadu 603211, India
5Department of Internal Medicine, Sundaram Health Centre, Tamil Nadu 632102, India
The outbreak of novel coronavirus disease (COVID-19) caused by SARS-CoV-2 severely challenges the economic stability of countries around the globe. Immune responses induced by active infection and vaccination have resulted in severe complications like thrombosis and cardiac injury in a measurable number of COVID-19 cases. Hence, knowledge of dissemination of these events is of clinical importance.Several bioactive enzymes are involved in the entry, replication and assembly of SARS-CoV-2 virus in host cells, of which angiotensinconverting enzyme-2 (ACE2) plays a crucial role in mediating the pathogenicity of SARS-CoV-2. ACE2 is expressed in multiple organs including the lungs, heart, digestive tract, liver, brain, eyes,pancreas and kidney, etc. Other than the lungs, the expression of ACE2 is found immensely on pericytes, cardiomyocytes, cardiac fibroblast, and epicardial adipocytes of the cardiac tissue. ACE2 mediates versatile pharmacological actions such as regulation of cardiac physiology, anti-oxidant, anti-fibrotic and anti-hypertrophic properties.
Recent studies hypothesise that SARS-CoV-2 infection reduces the expression of ACE2 at the membrane level[1]. In vitro studies demonstrated that ACE2 was downregulated in SARS-CoV-2 infected cell lines[2]. It is well known that cardiomyocytes have a higher expression of ACE2 and hence any alteration in the level of ACE2 may have a negative impact on cardiac function. In COVID-19 patients, the condition of hypoxemia greatly fluctuates the dynamics of blood flow to the cardiac tissue, which preambles the crisis of necrosis. Depleted ACE2 doubles the chances of inflammation (cytokine storm) and subsequent thrombotic risk in COVID-19 patients[3].
In vivo studies showed that the decrease in ACE2 level proportionately increases the expression of C-X3-C motif chemokine ligand 1 (CX3CL1). CX3CL1 is a larger fractalkine protein in the family of chemokines that exist in two forms:a membrane bound and soluble form[4]. In addition to ACE2 depletion, release of cytokines like TNF-α and IL-1β aggravates the expression of CX3CL1 on the endothelial matrix. Overexpression of CX3CL1 hastens the recruitment of T-cells and monocytes which contributes to the release of IL-1, IL-6, IL-10 and TNF-α in the microvascular environment. CX3CL1 exhibits direct involvement in the activation of platelets that upregulates the expression of P-selectin. Release of P-selectin through sequential degranulation of platelets triggers the episode of endotheliopathy and also promotes clot formation. Being a multifactorial component, CX3CL1 is now considered as a potential biomarker in ascertaining the severity of the cases. Available clinical data strongly correlates that patients with severe COVID-19 symptoms are observed with a two-fold increase in CX3CL1 level than in milder groups[5].
Limited expression of ACE2, in turn, destabilises the renin
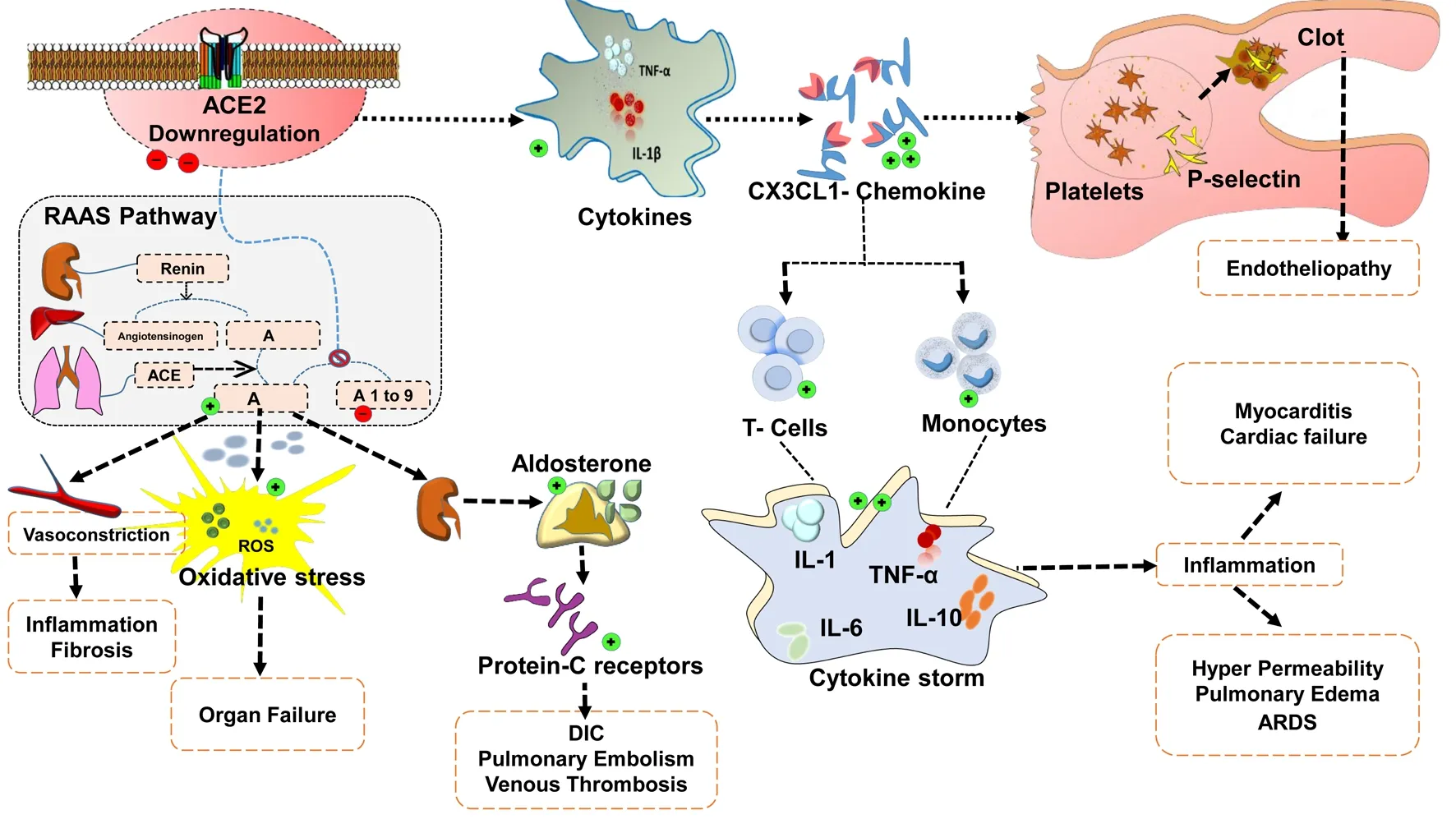
Figure 1. Downregulation of ACE2 and its pathological implications in the event of thrombosis and cardiac injury in COVID-19.
Ⅰ
Ⅱangiotensin aldosterone system mechanism, which leads to accumulation of angiotensin Ⅱ (AⅡ). Profound increase in AⅡprimarily responsible for vasoconstriction and for the production of reactive oxidative species. Reactive oxygen species are unstable moieties that quench the lipoidal layer of the healthy cells and thereby promote organ failure. Release of aldosterone guided by overwhelming AⅡ further upregulates the protein-C receptors which mediate hypercoagulable thrombotic events like DIC, pulmonary embolism and venous thrombosis in COVID-19 patients[6], as elucidated in Figure 1.A drastic sweep in cytokine storms is one of the major limitation factors observed in the SARS-CoV-2 infection. Massive overproduction of certain inflammatory cytokines upsurge the probability of cardiovascular disease such as hypotension, left ventricular hypertrophy and tachycardia. Clinical recommendation of renin angiotensin aldosterone system inhibitors may halt the progression of thrombotic events, meanwhile, therapeutics that selectively block CX3CL1 type chemokines may widen the scope of alternate strategy in the treatment of COVID-19 patients.Angiotensin Ⅱ receptor blocker (ARB) like losartan reveals a higher level of safety in patients with COVID-19 associated respiratory failure[7]. Results of three tire cohort clinical study shows that treatment with ARB’s exhibit significant clinical improvement in elderly COVID-19 patients[8].
Data from preclinical study emphasise that administration of angiotensin-converting enzyme inhibitor (ACEI-lisinopril) in rats upsurge the expression of cardiac ACE2 along with decreased plasma AⅡ level. Comparatively treatment with losartan (ARB)documents increased expression of cardiac ACE2 and its related activity in the experimental animals[9]. Outcome of meta-analysis over 1 664 (101 949 patients) published records acknowledge the protective benefits of using ACEIs and ARBs among COVID-19 patients with hypertension[10]. Despite clinical recommendations,large scale randomised trials are mandated to confirm the clinical implication of availing ACEIs and ARBs therapy in the treatment of COVID-19.
Conflict of interest statement
The authors declare no competing interests.
Acknowledgements
The authors thank the Indian Council of Medical Research (ICMR),Government of India, New Delhi, India.Authors’ contributions
All authors contributed equally in preparation of this article.
杂志排行

Asian Pacific Journal of Tropical Medicine的其它文章
- Effect of short- and long-term immunization of recombinant disorganized muscle protein-1 (rDIM-1) against human filarial parasite Brugia malayi in rodents
- Plasmid DNA encoding neutralizing human monoclonal antibody without enhancing activity protects against dengue virus infection in mice
- Spatio-temporal history of H9N2 viruses in Iran and neighbor countries by Bayesian analysis and molecular characterization
- Antibacterial resistance patterns of Acinetobacter baumannii complex: The results of Isfahan Antimicrobial Resistance Surveillance-1 Program
- Phylogeny of Brucella abortus strains isolated in the Russian Federation