Spatio-temporal history of H9N2 viruses in Iran and neighbor countries by Bayesian analysis and molecular characterization
2021-07-20NimaGhalekhaniSaiedBokaieSanaEybpooshHesameddinAkbarein
Nima Ghalekhani, Saied Bokaie, Sana Eybpoosh, Hesameddin Akbarein
1Divisions of Epidemiology & Zoonoses, Department of Food Hygiene and Quality Control, Faculty of Veterinary Medicine, University of Tehran, Iran 2HIV/STI Surveillance Research Center, and WHO Collaborating Center for HIV Surveillance, Institute for Futures Studies in Health, Kerman University of Medical Sciences, Kerman, Iran
3Department of Epidemiology and Biostatistics, Research Centre for Emerging and Reemerging Infectious Diseases, Pasteur Institute of Iran, Tehran,Iran
ABSTRACT
KEYWORDS: Influenza virus; H9N2; Bayesian phylogeographic;Iran
1. Introduction
Avian influenza is an important public health threat worldwide[1].Influenza A viruses are classified based on their hemagglutinin(HA) and neuraminidase (NA) surface proteins. So far, 18 HA and 11 NA subsets have been identified[2]. Based on their pathogenicity in birds, these strains are classified into low and highly pathogenic avian influenza. The compatibility and survival of these viruses in waterfowl made these birds natural reservoirs of the viruses. Birds can transmit the virus to a wide range of mammals, such as pigs,horses, and humans[3]. Among these strains, highly acute ones with H5 and H7 surface antigens and low pathogenicity strains with H9 have received more attention due to their potentials for causing pandemics through infected humans[3]. In 1999 and following in 2003, H9N2 (A/HK/2108/03) and (99/A/HK/1073) viruses were reported to infect humans, and since then, the H9N2 virus has been considered as a potential candidate for the development of new pandemics[3]. The H9N2 avian influenza virus is one of the major serotypes of influenza virus that has recently been distributed in different regions of the world[4].
Studies have shown that H9N2 is genetically similar to the H5N1 isolated from humans in 1997. Six genetic segments of H5N1 are shared with the H9N2 virus, which was found in a large number of Asian birds, and that virus had the potential to infect 18 people and kill six cases[5]. H9N2 viruses are classified into two major lineages,i.e., the “North American” and “Eurasian” lineages, circulating in poultry and wild birds. The Eurasian lineage is subdivided into three major lineages: a) Korean lineage [A/duck/Hong Kong/Y439/97(Y439-like) and A/chicken/Korea/38349-p96323/96 (Korean-like)];b) Y280 lineage [A/duck/Hong Kong/Y280/97-like (Y280-like), A/Chicken/Hong Kong/G9/97 (G9-like) and A/Chicken/Beijing/1/94(BJ94-like)]; and c) 3-G1 lineage [A/quail/Hong Kong/G1/97-like(G1-like)][5].
The H9N2 serotype was first isolated and identified in 1998 in Iran known as A/chicken/Iran/259/1998 in the Poultry Diseases Research and Diagnosis Department of Razi Institute and Faculty of Veterinary Medicine, University of Tehran[6]. Unfortunately, despite the initial plan to remove infected mother flocks and eggs, hatching and vaccination with inactivated vaccines, the disease outbreak spread rapidly through the country and has become endemic. It has caused a lot of damage to the country’s poultry industry[7].
Simultaneously with the spread of viral diseases such as influenza,the Bayesian phylogeographic method in 2009 was initiated and developed[8]. For example, Lemey et al. employed this method to investigate the global spread of influenza A (H5N1) and the rabies virus[8]. Several studies have also estimated the Spatio-temporal spread of some HIV-1 clades in different countries[9-11].
Despite the H9N2 avian influenza virus was mostly circulated in Iran and most of its neighboring countries, such as Pakistan,Afghanistan, Turkmenistan, Azerbaijan, Turkey, and Iraq, over the past decade[12-14], little is known about the virus’ circulation within and between these countries, the potential source of the epidemic in these countries, or its date of origin. To shed more light on the above knowledge gaps, we performed this Bayesian phylogeographic study on H9N2 genetic sequences available in the region. The results may be helpful in designing evidence-based control measures and prevention of onward virus transmissions.
2. Materials and methods
We downloaded all HA and NA nucleotide sequences of influenza H9N2 available up to December 25, 2020 from Iran and its neighboring countries (i.e., Pakistan, Afghanistan, Armenia, Turkmenistan,Azerbaijan, Turkey and Iraq) from the NCBI Influenza Virus Resource.Using information deposited in the NCBI Influenza Virus Resource,the data was annotated with sampling date and location, as well as host types (chickens, ducks, wild birds, etc.[15,16]. After the exclusion of identical sequences and isolates with unknown sampling time and location, a total of 268 HA and 211 NA sequences remained in our dataset. Multiple sequence alignment was performed using the ClustalW algorithm in MEGA v7.0 and manually inspected and trimmed for phylogenetic analysis[17]. The initial maximumlikelihood phylogenetic analysis was conducted using Hasegawa-Kishino-Yano for HA and General Time Reversible for N2 nucleotide substitution models, as identified by an online execution on the ATGC bioinformatics platform (http://www.atgc-montpelier.fr/). Analysis was performed with 1 000 bootstraps in MEGA v7.0[17].
We also performed a Bayesian Markov chain Monte Carlo(MCMC) method to infer the evolutionary dynamic and the most recent common ancestor for the HA and NA sequences[18]. Bayesian MCMC analysis was carried out using the BEAST v.2.5.241 package, with a Hasegawa-Kishino-Yano+G (HA sequences)and General Time Reversible+G+I (NA sequences) model as the substitution models along with an uncorrelated lognormal relaxed clock model and Bayesian skyline coalescent tree priors[19]. Two independent MCMC chains were run for 50 million generations(sampling every 5 000 steps) and were combined using the LogCombiner program v.1.54. Convergence was assessed based on the effective sampling size (ESS) after a 10% burn-in using Tracer software version 1.5. ESSs of 200 and more were accepted and a minimum ESS of 200 was required. The maximum clade credibility tree was generated in the TreeAnnotator program, while the initial 10% of trees were discarded as burn-in[18,20]. The maximum clade credibility tree was visualized in the FigTree program version 1.2.3[21].
Inferences about spatial dynamic and potential viral migration patterns of H9N2 within Iran and between Iran and its neighboring countries were made based on a discrete Bayesian phylogeographic model developed in BEAST software v.2.5.241[8]. The symmetric substitution model with the Bayesian Stochastic Search Variable Selection approach was performed in the SPREAD program v.1.0.7[22].
3. Results
3.1. Phylogenetic characteristics of H9N2 virus
The topology of the phylogenetic tree for the HA region suggested five genetic lineages. In the NA phylogenetic tree, we identified two lineages (Supplementary Figure 1 and 2). The biggest lineage in the HA tree is labeled as lineage 1 and consists of strains with Pakistan,Afghanistan, Iran, and Iraq origins. Chicken was the major host of the isolates within this lineage. The Lineage 4 is comprised only of strains that belong to Iran, but other lineages are formed by Pakistan and Iran’s strains (Figure 1 and 2).
The NA tree showed that the main locations of viruses in lineage 1 and 2 were Pakistan and Iran and for the NA tree chicken was the major host of the strains.
3.2. Phylogeography reconstruction of the H9N2 viruses in Iran and its neighboring countries
The basal branches of the HA tree were mainly occupied by Iran,with the root of the tree dating back to 1989. The virus later spread to Pakistan, most likely through a series of transmission events that began around 1992. The epidemic in Pakistan seems to be widely spread. The few available sequences from Iraq are dispersed independently throughout the tree, probably suggesting multiple independent clusters of infection between 1997 and 2008 and in 2013. Many sequences may not be available during this period(Figure 3).

Figure 1. Maximum likelihood phylogenetic tree of the hemagglutinin gene (Lineage 1).
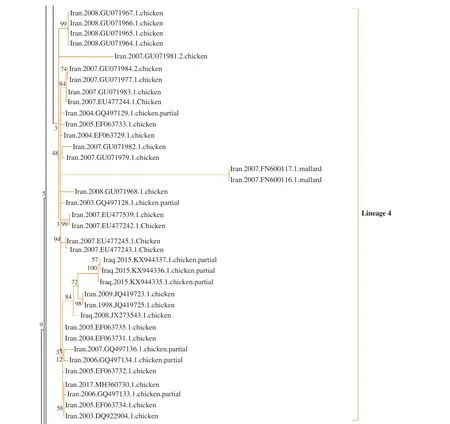
Figure 2. Maximum likelihood phylogenetic tree of the hemagglutinin gene (Lineage 4).
A new cluster was also discovered in Pakistan, which diverged from Iranian lineages or lineages from intermediate locations in 1995, but whose data was not included in the current study. Between 2004-2008 and around 2012, we observed multiple transmission events from Pakistan to Iran (Figure 3). The introduced lineages have experienced successful evolution and spread in Iran. The few sequences available from Afghanistan were nested within the Iranian lineages and dated back to 2014. Due to the small number of sequences available from Afghanistan and Iraq, it is not possible to provide a robust estimate of the date of origin as well as the magnitude of the outbreak in these countries (Figure 3).
Due to the high age and divergence of sequences in Iran and Pakistan in comparison with other countries in this sequence dataset,it seems that these two countries may have played a more highlighted role in the transmission pattern, but due to the insufficient sequences from other countries, conclusions about the role of the other countries may not be reliable.
Like the HA tree, the basal branches of the NA tree were also occupied by the sequences from Iran and Pakistan, respectively.The root height of the tree, which shows the estimated date of the epidemic’s onset in the region, was around 1992. The virus was first detected in Pakistan in 1996, and there appeared to be multiple backtransmission events from Pakistan to Iran in 2004, 2006, 2007, and 2010. The few available sequences from Afghanistan are nested within the Iranian branches, with a divergence date of 2008 and 2012(Figure 4).
The evolutionary rate for the HA and NA genes of the H9N2 viruses were estimated at 8.40×10[95% HPD: (6.34-10.00)×10]and 4.22×10[95% HPD: (3.07-5.52)×10] substitutions/site/year,respectively. The mean time of the most recent common ancestor of H9N2 viruses was 1988 (95% HPD: 1980-1995) for HA, and 1992(95% HPD: 1986-1996) for NA.
4. Discussion
To the best of our knowledge, this is the first Bayesian analysis of H9N2 viruses that attempts to fill the knowledge gap about the date

Figure 3. Maximum clade credibility phylogenies for the hemagglutinin gene of avian influenza A H9N2. The branches are colored according to the location of their nodes. The scale bar at the bottom indicates the years before the most recent sampling time (2019).

Figure 4. Maximum clade credibility phylogenies for the neuraminidase gene of avian influenza A H9N2. The branches are colored according to the location of their nodes. The scale bar at the bottom indicates the years before the most recent sampling time (2019).
of origin and circulation pathways of the influenza H9N2 virus in Iran and its neighboring countries. The results showed that the H9N2 epidemic may have started in Iran and Pakistan much earlier than the other investigated countries in the region, including Pakistan,Afghanistan, Armenia, Turkmenistan, Azerbaijan, Turkey, and Iraq.The onset of the H9N2 epidemic in the region was estimated to be between 1989 and 1992. An ongoing bidirectional dispersion of the virus between the investigated countries was also observed.
Our results showed that strains from the investigated countries were rooted in Pakistan and Iran. One justification for this may be the role of Pakistan and Iran in initiating the epidemic in the region.However, the abundance of sequence data available from Pakistan and Iran, and scarcity of data from other investigated countries,may have biased the topology of the phylogenetic tree. Lemey et al.discussed that sampling bias presents a critical challenge for popular discrete trait ancestral reconstruction procedures[23]. As further sequence data become available from currently under-represented countries, more rigorous conclusions could be made.
Molecular clock analysis estimates the onset date of the epidemic in Pakistan and Iran was around 1992 and 1989, respectively. This finding suggests that the case detection and reporting along with the influenza surveillance program in the initial years had some failures and the laboratory detection of these serotypes occurred several years after their introduction into these countries.
There was evidence of the virus moving in both directions, or bidirectionally, between Iran and Pakistan. A cluster in Pakistan showed divergence from Iranian lineages or intermediate locations whose data was not included in this analysis. The large genetic distance between Iran and Pakistan suggests that this transmission is probably an indirect transmission that caused an outbreak in Pakistan in 1995. Between 2004-2008 and around 2012, we observed multiple introduction events from Pakistan to Iran. The introduced lineages have experienced successful evolution and spread in Iran.
As small number of sequences were available from Afghanistan and Iraq, so the epidemic onset date could not be robustly estimated in these countries. The few available sequences from Iraq are dispersed independently throughout the tree, suggesting multiple independent introductions of the virus and multiple epidemic clusters probably occurred between 1997 and 2008 and in 2013. The few sequences available from Afghanistan were also nested within the Iranian lineages and dated back to 2014.
This circulation may be associated with the fact that Iran is one of the destinations for religious and recreational tourists and refugees from neighboring countries, especially from Iraq and Afghanistan,due to the low socioeconomic level and the civil war. This could have also contributed to between-country virus transmission where neighboring countries’ tourists and refugees might have exchanged the virus with the Iranians.
A phylogenetic study showed that H9N2 viruses isolated from birds in the UAE, Iran, and Pakistan were closely related to the H9N2 viruses that infected humans in Hong Kong[24]. In a broad phylogenetic study, all Iranian strains were located in one large cluster that also contained the sequences from neighboring countries,suggesting the epidemic in the region had originated from a common source[25]. A study by Toroghi et al. found a high homology between Iranian and German H9N2 viruses and suggested that the epidemic might have originated in Germany or in intermediate locations in Europe[25]. Detection of virus isolation from Pakistani parrots that were exported to Japan in 1998 was before the detection of the virus in Iran and with all these considerations it could be inferred that maybe Pakistan being infected before Iran[26,27]. The importance of ornamental birds in the spread of influenza viruses was then given more attention. Furthermore, given the high similarity of the NA and HA genes of the first Iranian influenza viruses isolated with the parakeet/chibal/1/97 virus, as well as the timing of virus isolation in Pakistan (1997, prior to the outbreak of Iranian avian influenza), the German origin of the epidemic in Iran may not be the case. Instead,the source of the epidemic in Iran may probably be the wildlife of its eastern countries, such as Pakistan[28]. The wide common land border between Iran and Pakistan and the intensive trade and transportation of poultry products, as well as the trade of ornamental birds, may support the latter hypothesis.
Furthermore, some of the region’s people’s traditions must be considered for their effects on virus transmission dynamics. Some people living near the common border between Iran, Afghanistan,and Pakistan engage in the live bird trade as a source of food. It was recently discovered that live bird markets with a lack of biosecurity measures may be linked to the accelerated spread of the H7N9 virus in China[29].
Therefore, regular surveillance of H9N2 viruses in the country,especially in the live bird markets, enhancing the biosecurity of the poultry industry and screening newly arriving immigrants and tourists from neighboring countries at the border should be considered to control the spread of the virus, which is a notable threat to public health and subsequent human infections have been reported[30]. Also, it is important that countries have a regional program in addition to domestic programs, and it will not be possible to control this disease without such a program. Furthermore, given the growing potential threat of the H9N2 virus to public health,surveillance of viral molecular evolution should be initiated. This is also critical for developing effective epidemic and pandemic prevention and control measures.
This study has some limitations. We could not deeply investigate the epidemiological parameters of the H9N2 virus in the neighboring countries like Iraq, Turkey, Armenia, Afghanistan and the Persian Gulf countries, due to the limited available sequence data. In addition, we could not directly estimate the evolutionary rates from the sequence alignments as the timespan of these datasets was not wide enough. Our model and dating estimates should be investigated further in the future as new sequences from additional countries and timespans become available, allowing for a more comprehensive evaluation of the H9N2 virus’s evolutionary history.
In summary, our results suggest that the H9N2 epidemic may have started in Iran and Pakistan much earlier than the other investigated countries in the region and was estimated to be between 1989 and 1992. Therefore, regular surveillance of H9N2 viruses in the country,especially in the live bird markets, enhancing the biosecurity of the poultry industry and screening newly arriving immigrants and tourists from neighboring countries at border, should be considered to control the spread of the virus. Also, it is important that countries have a regional program in addition to domestic programs and also surveillance of viral molecular evolution should be initiated. In the future, our model and dating estimates should be further investigated as new sequences from additional countries and timespans become available.
Conflict of interest statement
We declare that we have no conflict of interest.
Acknowledgements
We would like to express our gratitude to Institute for Futures Studies in Health, Kerman University of Medical Sciences,Kerman, Iran, especially Reza Sheikhzadeh (IT Manager) for his kind collaboration to access server computer in order to conduct computational analysis.
Authors’ contributions
N.G, S.B and S.E had made substantial contributions to design and perform the research. N.G and S.E had been involved in acquisition of data, or analysis and interpretation of data. NG and S.E had been involved in drafting the manuscript. N.G, S.B, S.E and H.A had been revised initial draft critically for important intellectual content and given final approval of the version to be published.
杂志排行

Asian Pacific Journal of Tropical Medicine的其它文章
- Effect of short- and long-term immunization of recombinant disorganized muscle protein-1 (rDIM-1) against human filarial parasite Brugia malayi in rodents
- Plasmid DNA encoding neutralizing human monoclonal antibody without enhancing activity protects against dengue virus infection in mice
- Antibacterial resistance patterns of Acinetobacter baumannii complex: The results of Isfahan Antimicrobial Resistance Surveillance-1 Program
- Phylogeny of Brucella abortus strains isolated in the Russian Federation
- ACE2 downregulation promotes thrombosis and cardiac injury in COVID-19 patients