Phylogeny of Brucella abortus strains isolated in the Russian Federation
2021-07-20DmitryKovalevDmitriyPonomarenkoSergeyPisarenkoNikolayShapakovAnnaKhachaturovaNataliaSerdyukOlgaBobryshevaAlexanderKulichenko
Dmitry A. Kovalev, Dmitriy G. Ponomarenko, Sergey V. Pisarenko, Nikolay A. Shapakov, Anna A.Khachaturova, Natalia S. Serdyuk, Olga V. Bobrysheva, Alexander N. Kulichenko
Stavropol Research Anti-Plague Institute, 355035, Stavropol, Russian Federation
ABSTRACT
KEYWORDS: Brucella abortus; Phylogeny; Evolution; Genome;SNP
1. Introduction
Brucellosis is an extremely dangerous, zoonotic infectious disease which may entail severe social and economic effects and is caused by a bacterium belonging to the Brucella genus including 12 species of microorganisms. The ones that are considered the most significant epidemiologically include Brucella (B.) melitensis, B.abortus and B. suis[1]. The typical host for B. abortus is cattle[2,3] and this Brucella species is to be found along with livestock basically all over the world.
In 2018, multilocus sequence typing data was used to show that the B. abortus species falls into 4 main clades[4], which correspond to the genetic lines of A, B, C1 and C2 as described by Whatmore et al[5]. At the same time, the data concerning the phylogeny of the B.abortus strains circulating in Russia is scarce.
This study aims at carrying out a whole-genome analysis of B.abortus strains isolated in the Russian Federation in order to identify their detailed position in the phylogenetic structure of the species global population, and to determine genetic relationships for isolates from different geographical areas.
2. Materials and methods
2.1. Bacterial strains
A total of 20 strains of B. abortus used throughout the study were obtained from the pathogenic microorganism collection of the Stavropol Anti-Plague Institute. Table 1 offers more detailed information regarding the biochemical properties of the isolates.
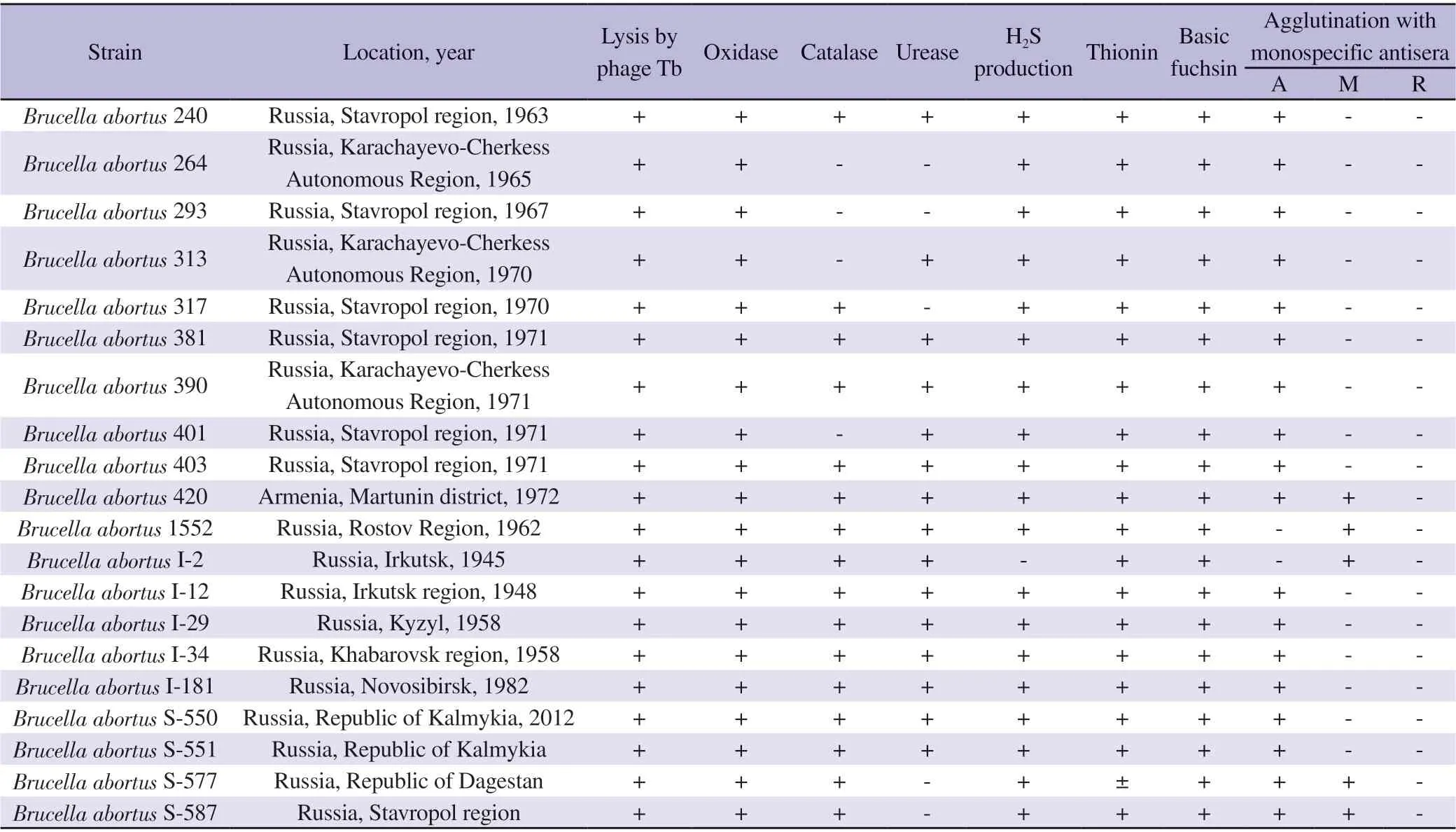
Table 1. Biochemical properties of Brucella abortus isolates.
2.2. Cultivation conditions and DNA extraction
The bacteria were cultured on Brucella agar for 48 h at a temperature of 37 ℃. The microbial suspension (concentration 2×10m.sub./mL) was decontaminated through adding sodium merthiolate to a final concentration of 0.01% and was further incubated at 56 ℃ for 30 min. Genomic DNA was isolated from 0.5 mL of decontaminated microbial suspension using PureLink Genomic DNA Kits (Life Technologies, USA). The concentration of the genomic DNA was determined with the Qubit 2.0 fluorimeter and the Qubit dsDNA HS Assay Kit (Invitrogen, Life Technologies, USA). The genomic DNA purity was evaluated with the NanoDrop 2000 spectrophotometer (Thermo Scientific, USA). All the manipulations with active strains were performed in a Level 3 biosafety laboratory.The obtained DNA preparations were stored at -20 ℃ until further use.
2.3. DNA sequencing
Genomic libraries with a ~400 bp read length were prepared using the Ion X PressTM Plus Fragment Library Kit (Life Technologies,USA) following the manufacturer’s protocol. Genome sequencing was done with the Ion Torrent PGM sequencer and the Ion 318 Chips Kit V2 chips (Life Technologies, USA). Once the quality assessment and data filtering procedures were complete, the resulting reads were collected into incomplete genome projects using SPAdes 3.12.0[6]. The S19 B. abortus strain genomic sequence (GeneBank:NC_010742.1, NC_010740.1) was used as a reference to assess the accuracy and efficiency of the genomic project assembly. The genome projects were annotated using the NCBI Prokaryotic Genome Annotation Pipeline.
2.4. Whole genome SNP analysis
Multiple genome alignment of 210 strains was performed in the REALPHY 1.10 software program[7] on default settings. To construct the core genome, 20 B. abortus genomes that we had sequenced were used, as well as 190 openly available B. abortus genomic sequences including complete genomes and genomic projects. The SNP search and identification in the core genome was carried out in the Mega 10 software program[8]. The site database of Brucella strain nucleotide polymorphisms is presented in Supplementary Table S1.
2.5. Phylogeographical and evolutionary analysis
The BEAST 2.3.0 software package was employed to study the phylogeographic taxon distribution based on whole-genome SNP analysis of B. abortus strains[9].We used the known B. abortus strain isolation dates to ensure the phylogenetic tree dating accuracy. The evolutionary model parameters were identified using the Jmodeltest 2 software[10], whereas Bayesian Markov chain Monte Carlo (MCMC)analysis was performed relying on the general time reversible +I+G(T+I+G) and the strict clock model. Three independent runs were held, with a chain length of 250 000 000 as per each run, and with a recording rate of every 1 000 generations. The convergence of the MCMC topology and parameters was evaluated in the Tracker 1.6 software program[11]. The trees were combined through the TreeAnnotator component from the BEAST 2.3.0 software package,thus obtaining a consensus tree, while the burn-in parameter for each chain was set at 20%. The 95% HPD Interval (confidence interval)value for the clock rate parameter was 5.678 8×10-9.273 7×10.
3. Results
3.1. General results
The genomic sequences of the 20 B. abortus strains isolated in Siberia and in South European Russia were obtained via highperformance sequencing using the IonTorrent PGM platform(Life Technologies, USA). The generated readings were collected based on a de novo approach into incomplete genomic projects.The genomic projects annotation was performed using the NCBI Prokaryotic Genome Annotation Pipeline. The resulting genomic projects were deposited in the GenBank database. Table 2 offers a view at the general features of the genomes.The genomic sequences of 258 B. abortus strains were used here,including 20 genomes of isolates obtained through this study as well as 238 genomic sequences from the GenBank international database.We used all the complete genomes and whole genome shotgun(WGS) projects that were available at the time the study was held. It was a close phylogenetic relationship described previously between B. abortus and B. melitensis species[7], which served the determining factor for taking the genomic sequence of the B. melitensis 16M strain as an external group. The data on the genomic sequences of the strains used through the work can be seen from Supplementary Table S2.We used REALPHY 1.10 with default settings to construct the multiple alignment matrix of the strain genomes[12]. The algorithms implemented in REALPHY allow obtaining a multiple genome alignment matrix that contains only orthologous nucleotide sequences (core genome). Some paralogous sequences are excluded from multiple alignment. The resulting multiple alignment matrix of complete genomes was used to construct the B. abortus phylogeny.The phylogenetic reconstruction was done using the BEAST 2.3.0 software package[9].In order to increase the phylogenetic tree dating accuracy, the core genome matrix had all those strain sequences excluded, where we failed to reliably identify the isolation date. All further manipulations,including phylogenetic and evolutionary analysis, along with SNP analysis, were performed relying on an edited core genome matrix,which contained the sequences of 209 strains. A phylogenetic tree offering a description of the evolutionary relationships of the studied strains can be seen in Supplementary Figure S1. Figure 1 presents a fragment of the phylogenetic tree with evolutionary relationships of strains isolated in Russia.
3.2. Phylogenetic and evolutionary analysis
The resulting dendrogram is divided into 4 major genetic lineages in accordance with existing ideas about the structure of the global population of B. abortus (Figure S1). The basal branch is represented by strains from Asia. B. abortus strains 63/294 and 88/217, isolatedin Kenya and Mozambique, respectively, belong to clade A. Other African strains belong to clade B. A representative group, which includes most of the Russian strains, is assigned to clade C1. Clade C2 includes the main part of isolates from North and South America with the B. abortus 544 strain.

Table 2. Characteristics of Brucella abortus genomic projects.
The Russian strains entered two separate clades, including the basal branch and the C1 branch, which is common in Eurasia.
The formation of the basal branch of the phylogenetic tree dates back to about 13 thousand years ago. The genotype includes strains of two sub-genotypes. Subgenotype a is represented by a single strain from China (BCB013, 2010), for which 1 661 specific SNPs were identified. Subgenotype b, differed by 579 SNP from other genotypes of the species, includes strains isolated in China(BCB027, 1983), in Armenia (420, 1972), in Russian Siberia (I-2,Irkutsk, 1945) and in South European Russia (C-587, Stavropol Territory, 2015; C-577, Republic of Dagestan, 2015). In general,805 specific SNPs were identified for the strains of this genotype.Comparison within subgenotype b genomes revealed 1 331 SNPs.
Based on the results of the analysis, the C1 genotype must have developed around the second half of the 6th Century BC. The genotype includes four subgenotypes marked as C1a-C1d. We found 115 clade-specific SNPs for the specified genotype strains.
The C1a subgenotype, which separated in the middle of the 4th Century AD, includes strains isolated in Russia, Greece and Italy (96 specific SNPs). When comparing the strains within the genotype,345 SNPs were described.
The subgenotype C1b subgroup, which includes strains isolated in Bolivia, India, Bangladesh, and Thailand, demonstrates the phylogenetic tree topology described earlier[13]. Besides, the subgenotype includes a strain from Russia (I-181, Novosibirsk,1982), as well as strains from Germany, France and the Great Britain. The C1b subgenotype differs from other genotypes and subgenotypes by a set of 96 SNPs. While comparing strains within the subgenotype, 622 SNPs were identified.
The extensive subgenotype C1d includes strains from Russia (I-29,I-12, 82, 240) and Italy (11796). The strains from Georgia made up a common subclade with the strain from Russia (317). A separate group was made up by strains from Russia (313), Egypt, Spain, Italy and Portugal. It is notable that most of the studied strains isolated in the south of Russia’s European part (Stavropol Territory, Republic of Kalmykia, Rostov Region) made up one subclade with strains from China and Mongolia. A set of 100 specific SNPs was detected for the C1d subgenotype. Comparison of the strains within the genotype allowed detecting 1 011 SNPs.
4. Discussion
The epidemiological situation connected with brucellosis in the Russian Federation over the past 10 years can be described as unfavorable featuring a decreasing trend in the incidence rate. Within the period of 2011-2020, there were 3 507 cases of brucellosis newly registered among people in Russia. The average long-term number of cases is 350 cases per year, including 28 cases among children aged below 17. The average long-term intensive morbidity rate as per 100 thousand people made up 0.24, children under 17 accounting for 0.1 of this value[14,15]. The highest number of brucellosis cases affecting people was observed in the North-Caucasus Federal District(NCFD)-2 291 (65.3% of the total number of brucellosis cases in Russia through 2011-2020) and the Southern Federal District (SFD)-503 cases (14.3%).
The basal branch of the resulting phylogenetic tree, which separated about 13 thousand years ago, includes two subgenotypes.Subgenotype a is a strain isolated in China, whereas subgenotype b involves strains from China, Russia, and Armenia. The subgenotype b strain from China (BCB027) as well as the group of strains isolated in Siberia (I-2), European Russia’s southern part (S-577, S-587)and Armenia (420) are likely to share a common origin, and must have diverged around 5 thousand years ago (3 000 BC) in the early Bronze Age. Within that period, large local blocks of communities of Eurasia’s population developed, which were involved into active interaction. The main two blocks of human communities, which were mostly engaged in agriculture and animal breeding, were located in the south of the central Alpine-Himalayan mountain belt:Sayans-Altai-Pamir and Tien-Shan-Caucasus-Carpathians-Alps.In the northern part of the Eurasian steppes, large nomadic tribes of cattle-raisers developed, while later on, communities of settled cattle-breeders emerged in the steppes of Eurasia[16].
To confirm the existence of the described genetic line B. abortus from Asia requires additional verification of the species affiliation of strains, as well as a study of a more representative sample of isolates from this region.
The obtained data suggests that the “African” genotypes A and B have not actually reached global spread and, as before, circulate mainly on one continent, which is consistent with the data from previous studies[5]. In recent decades, Africa has demonstrated a persistent trend to spread brucellosis (the pathogen migration) from the north of the Mediterranean Sea southwards the south into the continent[17]. There has been an increase in the number of human brucellosis cases registered in the countries located in the central and eastern parts of the continent. The disease cases in humans are mostly associated with the consumption of raw milk and unpasteurized dairy products from cattle[18]. At the same time, nearly each year the EU countries register brucellosis cases among refugees coming from Africa and the Middle East[19].
The C1 genotype isolates, on the other hand, are extremely common from Portugal in Western Europe to Thailand in Southeast Asia. The deviation of the C1 genotype tree branch may have occurred around the second part of the 6th Century BC. The strains of this genotype can be assumed to have originated from a common ancestor of Mediterranean origin, while their spread across Europe,North Africa and the Middle East may have occurred during the Roman conquests. Proof to this hypothesis can be seen from the geographical distribution of the C1a-C1c subgenotype strains isolated in Russia, Italy, Greece, Germany, France, Great Britain, as well as in Uganda, Iraq and a number of Asian countries.
The B. abortus I-34 strain isolated from an aborted cow fetus in 1958 in the Khabarovsk Territory of Russia, definitely belongs to a separate branch of the C1a subgenotype, which also includes isolates from Greece and Italy. The availability (circulation) of closely related brucellosis pathogen strains can be explained through their penetration from Asia together with brucellosis-contaminated cattle and further advance in the areas during the period of active trade going on between East and West, including that along the Great Silk Road from China to Rome through Persia, Parthia and the Middle East, which is a connection between East Asia and the Mediterranean(China and the Far East to the Middle East and Europe)[20,21].
The B. abortus I-181 strain isolated from human blood in 1982 in Novosibirsk (Russia) formed an individual C1b subgenotype branch. Apart from this strain, the subgenotype in question includes two branches: “South Asian” (India, Bangladesh, Thailand) and“European” (Germany, France, Great Britain). The significant genetic heterogeneity of this group of strains can be seen from 622 differentiating SNPs. One of the possible reasons behind the isolation of the C1a and C1b subgenotype strains in Siberia in the second part of the 20th Century could be the post-war practice of forced migration from rural areas together with livestock, including to the Krasnoyarsk Territory, Kemerovo and Irkutsk Regions,from the Baltic Republics, Western Ukraine as well as from the Caucasus[22]. Another factor that cannot be excluded is that Brucella strains may have been brought to Siberia, and then further to the East European plain, in the 1st Century BC-the time when trade between Asia and Europe was emerging, and the caravan routes ran from China to Siberia, including through the area currently known as the Trans-Baikal Territory, the Republic of Buryatia, the Irkutsk Region,the Krasnoyarsk Territory and the Novosibirsk Region[20,21].
The Eastern Mediterranean countries established active trade with Western Europe including that involving livestock products.Following the collapse of the Western Roman Empire, Mediterranean goods turned to be coming in specifically large supplies from the southwestern parts of Asia (mostly from the territory of present-day Turkey), Syria, and Egypt, while the areas in question featured poor status in terms of brucellosis contamination. At the same time, as feudalism was at its peak, from the 11th Century on, Western Europe was most active purchasing “eastern goods”, including high-value animals and raw products[21].
The time of the extensive C1d subgenotype separation dates back to the early 8th Century. Within our study, this subgenotype is represented by 53 strains, 19 of them isolated in Russia. On the phylogenetic tree, the C1d subgenotype is represented by three large clades. The first clade, which separated from the main branch around the year 1630 during the military conflicts between Eastern Georgia,seeking its independence, and Turkey and Iran[23], embraces 7 strains isolated in Georgia along with a strain from Russia (Stavropol Territory, 1970). The second clade consists of strains from Italy,Spain, Portugal and Egypt, as well as a separate strain isolated in Russia (Karachay-Cherkess Republic, 1970). The third clade,which emerged in the middle of the 17th Century, includes a group of 7 strains of Mongolian and Chinese origin, as well as a group of strains isolated in the south of the European part of Russia from 1962 to 2018. The period of intensive strain diversification in the North Caucasus coincides with the time when Russian was involved in the lengthy and bloody war in the Caucasus (Circassian war;1763-1864) [24].
Strains isolated in different parts of Russia (I-12, I-29, 82, 240), as well as strain 11796 from Italy, too, belong to the C1d subgenotype.Unfortunately, we could not obtain data regarding the biovaraffiliation of all the studied strains. It is obvious, though, that a particular strain belonging to one of the genotypes/subgenotypes based on wgSNP offers no clear connection with its biovar. The members of biovars 1 and 3, for instance, are assigned to different subgenotypes of the main genotypes. The discrepancy in the biovar affiliation and the position on the phylogenetic tree is accounted for by the fact that the biovar classification of B. abortus strains does not offer a full reflection of their genetic relationship or by possible inaccuracy in the biochemical determination of biovars[25].
In view of the above, the study can be seen as the first description for genomes of B. abortus strains circulating in Russia, to the best of our knowledge. Results of this study revealed a high degree of similarity among the whole-genome SNP profiles of B. abortus strains circulating within the same area, which allows employing the whole-genome SNP analysis as an effective tool used to identify the origin of certain individual isolates through epidemiological research.The expansion of knowledge concerning the unique features of B. abortus isolates obtained in different countries, as well as the integration of the respective data into international databases would allow better use of whole-genome sequencing data used to identify the genetic link degree between different strains; to identify the geographical region of the infection origin; to detect the causes, the conditions of brucellosis occurrence in animals and humans, as well as to localize the infection focus.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Acknowledgements
We thank all departments and institutes for the Brucella genome sequences that were used in this study: National Center for Biotechnology RSE, Beijing Institute of Biotechnology, Beijing Institute of Disease Control and Prevention, Broad Institute, Central India Institute of Medical Sciences, China Agricultural University,China Animal Disease Control Center, China Institute of Veterinary Drug Control, the Colombian Corporation for Agricultural Research, Inner Mongolia Agricultural University, Indian Veterinary Research Institute, Istituto Zooprofilattico Sperimentale del Mezzogiorno, Madurai Kamaraj University, National Institute for Communicable Disease Control & Prevention, Chinese Center for Disease Control and Prevention, National Center for Disease Control and Public Health, Scientific Centre for Expert Evaluation of Medicinal Products of the Ministry of Health of the Russian Federation, Sardarkrushinagar Dantiwada Agricultural University,Sardarkrushinagar Dantiwada Agricultural University, Shanghai JiaoTong University School of Medicine, University of New Hampshire, University of Sharjah, Virginia Bioinformatics Institute,the Wellcome Trust Sanger Institute.Authors’ contributions
DAK has developed a project and a research plan. DAK and ANK compiled the manuscript. DGP, AAK and NSS conducted bacteriological studies. SVP and OVB performed sequencing,genomes assembly and annotation. DAK, SVP and NAS conducted phylogenetic analysis. All authors have read and approved the final manuscript.
杂志排行

Asian Pacific Journal of Tropical Medicine的其它文章
- Effect of short- and long-term immunization of recombinant disorganized muscle protein-1 (rDIM-1) against human filarial parasite Brugia malayi in rodents
- Plasmid DNA encoding neutralizing human monoclonal antibody without enhancing activity protects against dengue virus infection in mice
- Spatio-temporal history of H9N2 viruses in Iran and neighbor countries by Bayesian analysis and molecular characterization
- Antibacterial resistance patterns of Acinetobacter baumannii complex: The results of Isfahan Antimicrobial Resistance Surveillance-1 Program
- ACE2 downregulation promotes thrombosis and cardiac injury in COVID-19 patients