基于生物信息学的甘薯基因组学等研究进展
2021-06-30马仁罡孙健英李宗芸
马仁罡 孙健英 李宗芸
摘要: 甘薯是重要的粮食、工业原料和新型能源作物,同时具有较高的营养价值。近年来,随着生物信息学分析手段、二代测序技术的发展,甘薯的基因组学、转录组学、蛋白质组学、代谢组学研究取得了较大进展。本文主要综述了近年来基于生物信息学技术,甘薯及其近缘野生种在基因组学、转录组学、蛋白质组学和代谢组学等方面研究中的进展,为甘薯品种改良提供参考和借鉴,并展望了甘薯未来的研究方向。
关键词: 甘薯;生物信息学;基因组学;转录组学;蛋白质组学;代谢组学
中图分类号: Q78;S531 文献标识码: A 文章编号: 1000-4440(2021)02-0531-08
Abstract: Sweet potato is an important food, industrial raw material and new energy crop with high nutritional value. In recent years, with the development of bioinformatics and the next generation sequencing technology, great progress had been got in the study of genomics, transcriptomics, proteomics and metabonomics of sweet potato. This paper mainly reviewed the research progress of genomics, transcriptomics, proteomics and metabonomics of sweet potato and its wild relatives based on bioinformatics in recent years. It can be used as a reference for the variety improvement of sweet potato. Finally, the future research direction of sweet potato is prospected.
Key words: sweet potato;bioinformatics;gen omics;transcriptomics;proteomics;metabonomics
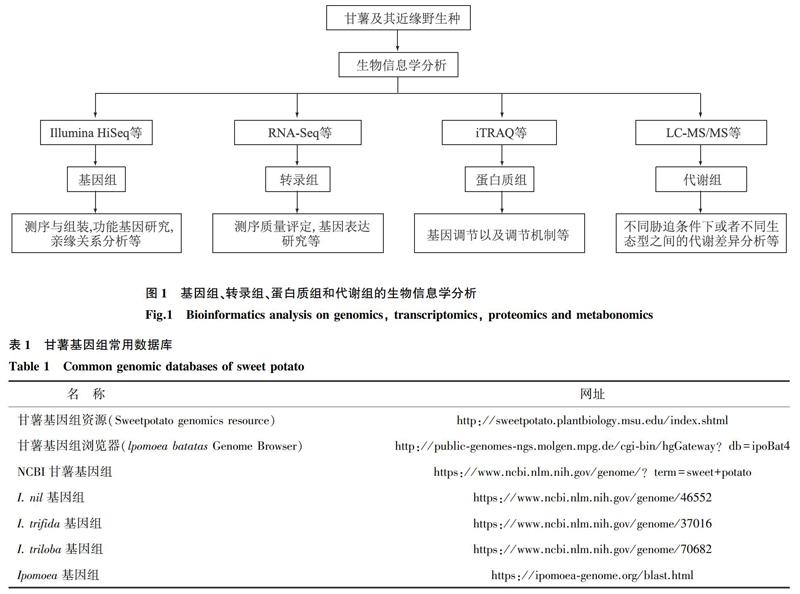
甘薯[Ipomoea batatas (L.) Lam]为旋花科(Convolvulaceae)番薯属(Ipomoea)植物,是世界重要的粮食、饲料、新型能源和工业原料作物,同时甘薯富含淀粉、花青素和硒等营养元素,是世界卫生组织推荐的最佳食品。因此,发展甘薯产业具有重要的战略意义。甘薯具有90条染色体(2n=6x=90),基因组大小为2 508 Mb[1]。与其他作物相比,甘薯基因组学相关研究难度较大,进展较为缓慢[2]。近几年来,生物信息学技术发展迅速,特别是新一代测序技术具有低成本和高效率的特点,促进了甘薯基因组、转录组、蛋白质组和代谢组的生物信息学分析(图1),并建立了一系列甘薯基因组的数据库(表1)。本文对近年来甘薯的基因组、转录组、蛋白质组和代谢组的研究进展进行了分析和总结,并以此为基础对甘薯研究进行了展望。
1 甘薯基因组研究进展
基因组学是一门交叉学科,一般分为以全基因测序和系统表征的结构基因组学[3],对基因功能研究的功能基因组学[4]和对基因组分析比较的比较基因组学[5]。
1.1 甘薯基因组测序和组装
近年来随着测序技术的不断发展,为了从近缘野生种中获取与生长发育和抗性相关的大量优异基因,提高甘薯育种技术,改善甘薯品质,并了解甘薯的起源和进化历程,研究者们对甘薯及其近缘野生种进行了基因组测序,包括核基因组测序以及质体基因组测序。
1.1.1 核基因組测序和组装 Ipomoea trifida(2x)是六倍体甘薯最有可能的二倍体祖先,为了辅助甘薯基因组的分析,Hirakawa等[6]使用Illumina HiSeq平台对I. trifida的2个品系(Mx23Hm和0431-1)进行了全基因组从头测序,也是第一次进行二倍体甘薯野生种全基因组从头测序。根据k-mer分布估算Mx23Hm和0431-1基因组大小分别为515.8 Mb和539.9 Mb,组装获得的核心序列为240.0 Mb和353.0 Mb,共鉴定出62 407个和109 449个可能的基因,大小分别为62.4 Mb和87.2 Mb,但此次组装并未达到染色体水平。直到2018年,报道了第一个关于I. trifida以及异源多倍体假说中甘薯祖先I. triloba的染色体水平的基因组测序[7],I. trifida株系NCNSP0306基因组大小为526.4 Mb,杂合度较高(0.24%),注释得到32 301个基因,I. triloba株系NCNSP0323的基因组大小为495.9 Mb,杂合度相对较低,注释得到31 426个基因。2019年,Li等[8]通过使用Illumina PE125对I. trifida var. Y22的基因组进行测序,生成了一个染色体水平基因组序列,基因组大小约为476.4 Mb,杂合度为2.2%,预测了30 227个可能的基因,其中79.76%被转录组数据所验证。
除了I. trifida和I. triloba,同属I. nil的基因组测序组装也已完成。2016年,Hoshino等[9]使用二代和三代测序技术对I. nil进行了测序,利用PacBio reads从头组装后基因组大小约为736.4 Mb,contig N50为1.87 Mb,scaffold N50为2.88 Mb,91.42%的组装序列被锚定在15条假染色体上,是旋花科中第一个组装到染色体水平的基因组。高质量I. nil基因组的公布也促进了甘薯基因组学的研究,Yang等[1]开创了一种基于二代测序序列的单倍型组装策略,对泰中6号进行全基因组从头组装,组装后1个染色体组大小约为836.3 Mb,scaffolds N50约为200.7 Kb,以I. nil基因组为参考基因组,有75.5%的组装序列锚定在15个假染色体上。
此外,一些甘薯近缘野生种的基因组特征信息也被报道,为这些物种全基因组深度测序提供了参考信息。马鞍藤(I. pes-caprae)基因组[10]初步测序分析预估其基因组大小为1 041.65 Mb,重复序列所占比率为74.52%,杂合度为0.99%;I. littoralis的基因组[11]大小预估为676.27 Mb,重复序列占比为60.98%,杂合度为0.81%;I. cordatotriloba基因组[12]大小约为539.69 MB,重复序列比例57.93%,杂合度0.40%。
基因文库的构建对甘薯基因组研究起到重要作用,颜朗等[13]为徐薯18构建180 bp、500 bp和2 Kb文库,进行从头组合式组装,预测甘薯基因组大小为2.7 Gb,重复序列比例为88%,挖掘出93 162条蛋白质编码基因序列,建立了具有创新性的从头组合式组装策略,为甘薯育种和基因组研究奠定了基础。第一个甘薯细菌人工染色体(Bacterial artificial chromosome,BAC)文库于2016年构建[14],此BAC文库包含240 384个克隆,平均插入大小为101 kb,基因组覆盖度7.93~10.82 X。
1.1.2 质体基因组测序和组装 甘薯及其野生种的叶绿体基因组结构为典型的四分体结构,包括2个反向重复区(Inverted repeats,IRs),一个大的单拷贝区(Large single copy,LSC)和一个小的单拷贝区(Small single copy,SSC)。Eserman等[15]对33个番薯亚族物种进行了叶绿体基因组测序,并基于叶绿体基因组序列对番薯亚族物种进行了详细的系统发育分析。Yan等[16]对甘薯叶绿体全基因组进行了分析,结果表明甘薯叶绿体基因组包含145个基因,其中蛋白质编码基因为94个(单拷贝基因72个,双拷贝基因11个)。Sun等[17]对8个甘薯近缘野生种的叶绿体基因组进行了测序和组装,结果显示,这些近缘野生种叶绿体基因组长度为161 225~161 721 bp,含有80个蛋白质编码基因、4个rRNA 和37个tRNA基因。Park等[18]利用NextSeq平台对6个甘薯野生近缘种进行了叶绿体全基因组测序,基因组长度范围为161 354~161 750 bp,共鉴定出112个基因,含有78个蛋白质编码区,30个tRNA基因和4个rRNA基因。随着I. nil基因组的发表,研究者也公布了I. nil的线粒体和叶绿体基因组[9],它们的基因组大小分别为0.27 Mb和0.16 Mb,G和C含量分别为44.45%和37.47%。
以上研究结果不仅促进了我们对甘薯及其野生近缘种基因组特征的认识,为进行甘薯遗传改良提升育种品质提供了重要的遗传信息,还加速了甘薯系统发生和起源进化的研究。如Wu等[7]确定了贮藏根中与类胡萝卜素生物合成相关的基因和等位基因,这使高维生素A含量品种的高效育种成为可能;Li等[8]则发现了BMY11与甘薯贮藏根发育有关,可促进淀粉的积累和贮藏根的膨胀,为后期分子育种提供了研究基础。甘薯泰中6号、甘薯近缘野生种I. trifida和I. triloba基因组的测序和组装为甘薯不同起源和进化假说提供了基因组方面的证据。
1.2 遗传图谱构建
遗传图谱是甘薯分子育种的重要工具之一,构建甘薯遗传图谱在遗传演化分析和数量性状位点(Quantitative trait locus,QTL)基因定位研究中起到重要作用[19]。随着测序技术的发展,遗传图谱构建所使用的技术从相对落后的基于聚合酶链式反应(Polymerase chain reaction,PCR)的分子标记技术发展到基于高通量测序的分子标记技术。
1.2.1 基于PCR技术的遗传图谱构建 李爱贤等[20]以漯徐薯8号和郑薯20杂交产生的后代为研究对象,利用序列相关扩增多态性(Sequence related amplified polymorphism,SRAP)技术和JoinMap 3.0软件构建了分子连锁图谱,漯徐薯8号连锁图谱有473个SRAP标记组成了81个连锁群,总图距为5 802.46 cM,平均标记间距为10.16 cM。郑薯20连锁图谱由328个SRAP标记组成了66个连锁群,总图距为3 967.90 cM,平均标记间距为12.02 cM。揭琴等[21]也利用JoinMap 3.0软件,结合扩增片段长度多态性(Amplified fragment length polymorphism,AFLP)标记,构建了徐781和徐薯18的分子连锁图谱,均含有90个连锁群,分别含有1 878和1 868个AFLP标记,总图距为8 214.0 cM和8 319.0 cM,平均标记间距为4.4 cM和4.5 cM。Kim等[22]使用表达序列标签简单重复序列(Expressed sequence tag simple sequence repeat,EST-SSR)標记技术,以Yeseumi和Annobeny杂交产生的后代为研究对象,共开发了245个EST-SSR标记,使用Mapmaker 3.0软件进行标记分组,并使用Mapchart 2.2软件利用其中210个标记进行甘薯遗传图谱的构建,构建出的图谱总长为1 508.1 cM,平均距离为7.2 cM。
1.2.2 基于高通量测序的遗传图谱构建 随着测序技术的发展,产生了基于高通量测序的遗传图谱构建方法,与基于PCR的分子标记技术相比,具有工作量更小,成本低,准确性高以及可重复等优点。Shirasawa等[23]通过徐薯18的自花授粉产生了S1定位群体,利用限制性双酶切位点关联DNA测序技术(Double digest restriction-site associated DNA sequence,ddRAD-Seq)对此群体文库构建和测序分析,共得到28 087个单核苷酸多态性(Single nucleotide polymorphism,SNP)位点,可映射到96个连锁群上,总距离为33 020.4 cM,分析得出,国内品种间遗传距离较近,国内品种与国外品种遗传距离较远。同样,Su等[24]选用264~314 bp的DNA片段作为特异性位点扩增片段(Specific-locus amplified fragment,SLAF),使用Illumina Hiseq 2500进行测序,利用62 363个SNP将197个来自全球各地的样本分为3组,各组材料间遗传距离值为0.290~0.311,平均多态性信息含量均为0.232~0.251,最小等位基因频率为0.207~0.222。李慧峰等[25]通过简化基因组测序技术对122份甘薯种质资源进行SNP位点开发并对种质资源群体结构和遗传进化进行分析,共获得基因组大小为563.18 Mb,G+C含量平均为38.17%,共开发出2 388 759个高质量的SLAF标签,平均深度为17.45 X,其中含有3.26%的多态性SLAF标签,共77 761个,含有129 063个群体SNP位点。Mollinari等[26]利用MAPpoly软件,构建了第一个六倍体甘薯的多位点整合遗传图谱,最终图谱包含30 684个SNP标记,全长2 708.4 cM。此外,Sasai等[27]将几种标记技术组合起来,对J-Red和Choshu种间杂交产生的F1后代结合SSR标记、SNP标记以及逆转录转座子插入多态性分析,建立了高密度连锁图谱,J-Red连锁图谱总长为13 247 cM,各连锁群平均长度为147.2 cM,标记间平均距离为2.09 cM,平均有70.5个标记。Choshu连锁图谱总长为12 241.8 cM,各连锁群平均长度136.0 cM,标记间平均距离2.0 cM,平均有66.8个标记。
以上研究结果不仅绘制了遗传图谱,还对甘薯的群体结构和遗传多样性进行了分析,为甘薯基因改良和甘薯育种提供了帮助。
2 甘薯转录组研究进展
转录组是指特定细胞或组织中全部转录产物,包括信使RNA,核糖体RNA、转运RNA以及非编码RNA[28],对转录组分析能够提供测序质量评定、信息定位、基因表达和可变剪接位点等信息[29]。
2.1 非生物胁迫相关转录组分析
转录组测序方面,Ding等[30]利用单分子实时测序和第二代测序技术,对甘薯和I. trifida的全长转录序列进行了测定,分别获得53 864个和51 184个高质量的长阅读转录本,覆盖了基因组中约10 439个和10 452个基因座,能够预测96.83%和96.82%的转录本的开放阅读框架,并识别出34 963个和33 637个全长cDNA序列,1 401个和1 457个转录因子,25 315个和27 090个简单重复序列,1 656个和1 389个长的非编码RNA,5 251个和8 901个可变剪接事件,促进了六倍体甘薯的比较和功能基因组研究。
在转录组测序过程中,研究者们发现了一些病毒和非生物胁迫相关信息。Jo等[31]使用RNA测序技术(RNA Sequencing,RNA-Seq)对韩国广泛栽培的2个甘薯品种Beni Haruka和Hogammi的10个不同文库进行测序分析,发现2种新病毒[Sweet potato virus E(SPVE)和Sweet potato virus F(SPVF)]感染,结合前人的研究,提供了一个完整的韩国甘薯可能会感染的病毒清单。Kuo等[32]通过Illumina MiSeq平台对转录组文库进行测序,利用trinity软件重新组装了contigs RNA序列,进行转录组分析,研究了甘薯中與创伤相关的miR408及其靶基因。Yang等[33]共鉴定出475个已知的miRNA和175个新的miRNA,并通过降解组测序验证了314个miRNA的636个靶基因,发现大部分miRNA在盐胁迫下的表达与其靶基因的表达呈负相关。Weng等[34]基于甘薯转录组数据库,鉴定了miR2111的靶基因IbFBK,通过试验发现miR2111的抑制作用导致IbFBK表达量的增加,并可能调节IbCNR8在损伤时的蛋白质降解。
在非生物胁迫下进行转录组分析有利于帮助我们筛选出与胁迫相关的基因,如Ji等[35]对低温胁迫下和恢复过程中的叶片进行了转录组从头测序,分别鉴定出2 461个和1 017个差异表达基因,同时研究了抗氧化酶途径介导的活性氧(Reactive oxygen species,ROS)响应的相关基因,进一步研究了甘薯对冷胁迫的响应机制。并在之后的研究中从头组装冷藏能力较强的徐薯15-1和冷藏能力较弱的徐薯15-4 2个甘薯株系的RNA-Seq数据[36],产生了27 636个基因,N50值为1 204 bp,共有525个差异表达基因,并筛选出抗寒的候选基因。干旱胁迫下,Lau等[37]使用RNA-Seq技术,从干旱处理下的甘薯中,筛选出122个耐旱候选基因。在盐胁迫下,Arisha等[38]建立了5个cDNA文库,进行转录组测序和分析,筛选出了新的耐盐胁迫候选基因。吴燕等[39]从甘薯的转录组数据中筛选出238个与耐盐、耐旱相关的基因。Sung等[40]通过分析甘薯在根结线虫感染过程中转录组的变化,确定了可能有助于预防甘薯块根根结线虫感染的候选基因。
2.2 发育相关转录组分析
转录组测序分析不仅可以发现与非生物胁迫相关的基因,还可以发现与发育相关的基因。块根发育方面,Dong等[41]为探讨甘薯贮藏根形成和发育的分子机制,以4个不同时期的须根中段和贮藏根为材料,制备了5个cDNA文库,通过Illumina HiSeq 2000测序平台,共鉴定出26 273个差异表达基因,根据对贮藏根形成和发育过程中表现出相似表达谱的基因进行聚类分析,确定了4个显著的基因亚群。Li等[42]则在不同甘氨酸处理的贮藏根中共鉴定出4 836个差异表达基因,其中涉及碳水化合物代谢的基因最多,为1 830个,并通过实时荧光定量PCR(qRT-PCR)证实外源甘氨酸通过加强光合作用和增加植物激素促进贮藏根的生长,通过加速碳水化合物的代谢和调控淀粉相关基因的表达,促进贮藏根淀粉的生物合成。He等[43]从徐薯18和徐紫薯3的块根中构建了小分子RNA和降解组文库,共鉴定出191个已知的miRNA、33个新的miRNA、180个靶基因和5个新的ib-miRNA,并筛选出与花青素相关的miRNA和相应的靶基因。Ponniah等[44]将甘薯与I. trifida转录组数据相比较,发现了一些甘薯特异的转录因子、蛋白激酶和家族蛋白质,提示这些基因可能在贮藏根的形成中起到重要作用,并筛选出一些分子标记,为进一步研究奠定了基础。
花发育方面,Tao等[45]分析比较甘薯花的转录组数据,筛选出了调控开花的相关基因。Wei等[46]对嫁接后的I. nil进行转录组测序分析,结合PacBioIso-Seq和IlluminaRNA-seq,分析了花期嫁接过程中的转录组变化,筛选出与花青素生物合成、光合作用和乙烯信号转导通路相关基因,帮助研究者们更好地认识了嫁接作用的分子机制,为甘薯育种奠定了基础。
3 甘薯蛋白质组研究进展
蛋白质组的研究在甘薯领域内相对较少,Almohanna等[47]采用2种互补的蛋白质提取方法和自动化的蛋白质组平台,分析了甘薯组织特异性的蛋白质组,成功鉴定出与4 321个非冗余蛋白质相对应的74 255个多肽,39 916个多肽定位于叶中3 143个特异的蛋白质上,34 339个多肽定位于根中2 928个独特的蛋白质上,总共预测了741个新的蛋白质编码基因,鉴定了甘薯叶片和贮藏根蛋白质组的组成和功能特征,为甘薯基因组功能注释研究做出了贡献。Dong等[41]利用同位素标记相对和绝对定量(Isobaric tags for relative and absolute quantification,iTRAQ)对不同时期的须根和贮藏根进行了比较蛋白质组分析,从5个文库中共鉴定7 727个蛋白质,其中,在基因本体(Gene ontology,GO)数据库的注释率为98.81%,蛋白质家族、域和作用位点信息整合数据资源(InterPro,IPR)数据库的注释率为88.08%,京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)数据库的注释率为87.03%,蛋白质相邻类的聚簇(Cluster of orthologous groups of proteins,COG)数据库的注释率为56.59%,与转录组研究结合,证明贮藏根的发育存在转录后调控,揭示了关于贮藏根形成的途径。
4 甘薯代谢组研究进展
代谢组学是系统研究生物体或生物体内小分子代谢产物的科学[48],在植物中的研究主要是对植物在受到外界影响前后代谢成分的定性、定量分析[49],也可以通过对代谢组数据的分析来区分不同表型内在代谢差异[50]。在研究中,代谢组多与基因组、转录组以及蛋白质组联合进行分析[51]。
在热处理过程中,甘薯的代谢产物会减少或变化,如4種在巴西生长的橙肉红薯经过热处理后,类胡萝卜素、酚类化合物的含量以及抗氧化能力都显著降低[52]。Rautenbach等[53]的研究结果也证明了这一点,热处理后的4个甘薯品种的类胡萝卜素和维生素C含量降低,且相比于橙肉甘薯,白肉甘薯降低的更多。Kim等[54]在热处理前后对12个甘薯品种的酚类化合物含量和抗氧化活性进行了比对,发现热处理后红薯的水杨酸、香草酸、五倍子酸和咖啡酸含量增加,原儿茶酸和绿原酸含量下降。
对不同薯肉颜色甘薯进行代谢组研究,可以分析不同薯肉颜色甘薯的代谢差异和营养成分。Teow等[55]通过比较19种不同薯肉颜色甘薯的抗氧化活性、酚类含量和β-胡萝卜素含量,总结出总酚含量可以作为甘薯抗氧化活性的指标,且试验中紫肉甘薯的抗氧化活性比白肉甘薯高。根据研究卢旺达种植的2个白肉和2个橙肉甘薯的水分、蛋白质、粗纤维、胡萝卜素和还原糖含量,推测橙肉甘薯更有营养,胡萝卜素只在橙肉甘薯中存在[56]。此外,对不同薯肉颜色甘薯的14个初级代谢产物和18个次生代谢产物分析结果显示,花青素只存在于紫肉甘薯中,且酚类和类黄酮含量高于其他肉色甘薯,橙肉甘薯的类胡萝卜素高于其他甘薯[57]。但在之后的研究中发现,紫肉甘薯P40的花青素却低于白肉甘薯Bonita和橙肉甘薯Beauregard,这一出乎意料的结果刷新了对甘薯中花青素含量的认知[58]。对不同薯肉颜色甘薯的类黄酮也开展了更深研究,共鉴定了213种代谢物,其中黄酮类化合物29种,酚酸类化合物27种,发现代谢差异的原因是薯肉颜色的不同,且与苯丙和黄酮的生物合成相关[59]。对薯肉颜色相同的甘薯进行研究,有利于选取适合种植的高营养物质含量的甘薯品种。如对5个紫肉品种甘薯的酚类化合物进行研究,可以区分各品种所富含的酚类化合物[60]。
5 展望
生物信息学和测序技术的发展[61-64]极大地促进了对甘薯的研究,目前已对部分甘薯品种及2个近缘野生种完成了测序和较高水平的组装。此外,通过对基因组和转录组的测序分析,筛选得到一些基因,并对一些已知的基因功能进行了验证。目前, 对蛋白质组和代谢组的研究也日益增多,对甘薯品种的选择提供了帮助。综上所述,近几年来生物信息学与基因组学等研究的结合,使得甘薯及其近缘野生种的基因组、转录组、蛋白质组和代谢组研究都取得了长足的进展。在未来的研究中,需进行深度测序和高质量的组装,加强对甘薯及其野生近缘种基因组特征的认识,绘制出更精细的遗传图谱,分析筛选出更全面的功能基因,为甘薯系统发生和进化研究提出更多证据,并通过对蛋白质组和代谢组的进一步研究,选择培育更有营养的甘薯品种,为甘薯育种奠定更为坚实的基础,为中国的甘薯产业做出贡献。
参考文献:
[1] YANG J, MOEINZADEH M, KUHL H, et al. Haplotype-resolved sweet potato genome traces back its hexaploidization history[J]. Nature Plants,2017, 3(9): 696-703.
[2] 李 强,刘庆昌,马代夫,等. 甘薯遗传转化研究现状、问题及展望[J]. 分子植物育种, 2005(1): 99-106.
[3] 薛方方,王义聪,杜 美,等. 高通量全基因组测序应用于淋球菌耐药的研究进展[J]. 中国艾滋病性病, 2020, 26(6): 671-672.
[4] WANG D, LI F, CAO S, et al. Genomic and functional genomics analyses of gluten proteins and prospect for simultaneous improvement of end-use and health-related traits in wheat[J]. Theor Appl Genet, 2020, 133(5): 1521-1539.
[5] TORRES-CORRAL Y, SANTOS Y. Comparative genomics of Streptococcus parauberis: new target for molecular identification of serotype III[J]. Applied Microbiology and Biotechnology, 2020, 104(14): 6211-6222.
[6] HIRAKAWA H, OKADA Y, TABUCHI H, et al. Survey of genome sequences in a wild sweet potato, Ipomoea trifida (H.B.K) G. Don[J]. DNA Research, 2015, 22(2): 171-179.
[7] WU S, LAU K H, CAO Q, et al. Genome sequences of two diploid wild relatives of cultivated sweetpotato reveal targets for genetic improvement[J]. Nature Communications, 2018, 9(1): 4512-4580.
[8] LI M, YANG S, XU W, et al. The wild sweetpotato (Ipomoea trifida) genome provides insights into storage root development[J]. BMC Plant Biology, 2019, 19(1): 119.
[9] HOSHINO A, JAYAKUMAR V, NITASAKA E, et al. Genome sequence and analysis of the Japanese morning glory Ipomoea nil[J]. Nature Communications, 2016, 7(1): 13295.
[10]霍恺森,赵冬兰,陈艳丽,等. 甘薯属耐盐植物马鞍藤基因组大小及特征分析[J]. 植物遗传资源学报, 2019, 20(3): 728-735.
[11]霍恺森,曹清河,王 珧,等. 甘薯近缘野生种Ipomoea Littoralis全基因组Survey分析[J]. 热带作物学报, 2019, 40(10): 2001-2005.
[12]王 珧,邓逸桐,戴习彬,等. 甘薯近缘种Ipomoea cordatotriloba基因组大小测定及高通量调查测序[J]. 热带作物学报, 2020,41(6):1154-1159.
[13]颜 朗,李雪丹,吴 燕,等. 甘薯基因组概览分析及重要功能基因挖掘[C].//中国遗传学会. 2015中国遗传学会大会论文摘要汇编: 北京:科学出版社, 2015: 42.
[14]SI Z, DU B, HUO J, et al. A genome-wide BAC-end sequence survey provides first insights into sweetpotato [Ipomoea batatas (L.) Lam.]genome composition[J]. BMC Genomics, 2016, 17(1): 945.
[15]ESERMAN L A, TILEY G P, JARRET R L, et al. Phylogenetics and diversification of morning glories (tribe Ipomoeeae, Convolvulaceae) based on whole plastome sequences[J]. American Journal of Botany, 2014, 101(1): 92-103.
[16]YAN L, LAI X, LI X, et al. Analyses of the complete genome and gene expression of chloroplast of sweet potato (Ipomoea batata)[J]. PLoS One, 2015, 10(4): e124083.
[17]SUN J, DONG X, CAO Q, et al. A systematic comparison of eight new plastome sequences from Ipomoea L.[J]. Peer J, 2019, 7: e6563.
[18]PARK I, YANG S, KIM W J, et al. The complete chloroplast genomes of six Ipomoea species and indel marker development for the discrimination of authentic pharbitidis semen (Seeds of I. nil or I. purpurea)[J]. Frontiers in Plant Science, 2018, 9: 965.
[19]胡志程,周梦迪,吕建春,等. 甜瓜遗传图谱与基因定位研究进展[J]. 分子植物育种, 2020, 18(7): 2290-2295.
[20]李爱贤,刘庆昌,王庆美,等. 利用SRAP标记构建甘薯分子连锁图谱[J]. 作物学报, 2010, 36(8): 1286-1295.
[21]揭 琴,李 华,翟 红,等. 甘薯抗莖线虫病基因AFLP标记的开发[J].农业生物技术学报, 2008,16(5): 837-841.
[22]KIM J, CHUNG I K, KIM K. Construction of a genetic map using EST-SSR markers and QTL analysis of major agronomic characters in hexaploid sweet potato [ Ipomoea batatas (L.) Lam][J]. PLoS One, 2017, 12(10): e185073.
[23]SHIRASAWA K, TANAKA M, TAKAHATA Y, et al. A high-density SNP genetic map consisting of a complete set of homologous groups in autohexaploid sweetpotato (Ipomoea batatas)[J]. Scientific Reports, 2017, 7(1): 44207.
[24]SU W, WANG L, LEI J, et al. Genome-wide assessment of population structure and genetic diversity and development of a core germplasm set for sweet potato based on specific length amplified fragment (SLAF) sequencing[J]. PLoS One, 2017, 12(2): e172066.
[25]李慧峰,黃咏梅,李彦青,等. 基于SLAF-seq技术的甘薯种质资源群体遗传进化分析[J]. 热带作物学报, 2019, 40(12): 2390-2396.
[26]MOLLINARI M, OLUKOLU B A, PEREIRA G D S, et al. Unraveling the hexaploid sweetpotato inheritance using ultra-dense multilocus mapping[J]. G3-Genes Genomes Genetics, 2020, 10(1): 281-292.
[27]SASAI R, TABUCHI H, SHIRASAWA K, et al. Development of molecular markers associated with resistance to Meloidogyne incognita by performing quantitative trait locus analysis and genome-wide association study in sweetpotato[J]. DNA Research, 2019, 26(5): 399-409.
[28]崔 凯,吴伟伟,刁其玉. 转录组测序技术的研究和应用进展[J]. 生物技术通报, 2019, 35(7): 1-9.
[29]宋尚桥,马围围,张超龙,等. 基于转录组测序生物信息学分析的研究进展[J]. 中国畜牧兽医, 2020, 47(2): 392-398.
[30]DING N, CUI H, MIAO Y, et al. Single-molecule real-time sequencing identifies massive full-length cDNAs and alternative-splicing events that facilitate comparative and functional genomics study in the hexaploid crop sweet potato[J]. Peer J, 2019, 7: e7933.
[31]JO Y, KIM S, CHOI H, et al. Sweet potato viromes in eight different geographical regions in Korea and two different cultivars[J]. Scientific Reports, 2020, 10(1): 2588.
[32]KUO Y, LIN Y, LI Y, et al. MicroR408 regulates defense response upon wounding in sweet potato[J]. Journal of Experimental Botany, 2019, 70(2): 469-483.
[33]YANG Z, ZHU P, KANG H, et al. High-throughput deep sequencing reveals the important role that microRNAs play in the salt response in sweet potato (Ipomoea batatas L.)[J]. BMC Genomics, 2020, 21(1): 116-164.
[34]WENG S, KUO Y, KING Y, et al. Regulation of micoRNA2111 and its target IbFBK in sweet potato on wounding[J]. Plant Science, 2020, 292: 110391.
[35]JI C Y, BIAN X, LEE C, et al. De novo transcriptome sequencing and gene expression profiling of sweet potato leaves during low temperature stress and recovery[J]. Gene, 2019, 700: 23-30.
[36]JI C Y, HO S K, LEE C, et al. Comparative transcriptome profiling of tuberous roots of two sweetpotato lines with contrasting low temperature tolerance during storage[J]. Gene, 2020, 727:144244.
[37]LAU K H, ROSARIO HERRERA M, CRISOVAN E, et al. Transcriptomic analysis of sweet potato under dehydration stress identifies candidate genes for drought tolerance[J]. Plant Direct, 2018, 2(10): e92.
[38]ARISHA M H, ABOELNASR H, AHMAD M Q, et al. Transcriptome sequencing and whole genome expression profiling of hexaploid sweetpotato under salt stress[J]. BMC Genomics, 2020, 21(1): 118-197.
[39]吴 燕,颜 朗,李雪丹,等. 甘薯耐旱和耐盐基因的挖掘和表达分析[J]. 四川大学学报, 2016, 53(5): 1147-1154.
[40]SUNG Y W, LEE I H, SHIM D, et al. Transcriptomic changes in sweetpotato peroxidases in response to infection with the root-knot nematode Meloidogyne incognita[J]. Molecular Biology Reports, 2019, 46(4): 4555-4564.
[41]DONG T, ZHU M, YU J, et al. RNA-Seq and iTRAQ reveal multiple pathways involved in storage root formation and development in sweet potato (Ipomoea batatas L.)[J]. BMC Plant Biology, 2019, 19(1): 136.
[42]LI C, YAO W, WANG J, et al. A novel effect of glycine on the growth and starch biosynthesis of storage root in sweetpotato (Ipomoea batatas Lam.)[J]. Plant Physiology and Biochemistry, 2019, 144: 395-403.
[43]HE L, TANG R, SHI X, et al. Uncovering anthocyanin biosynthesis related microRNAs and their target genes by small RNA and degradome sequencing in tuberous roots of sweetpotato[J]. BMC Plant Biology, 2019, 19(1): 232.
[44]PONNIAH S K, THIMMAPURAM J, BHIDE K, et al. Comparative analysis of the root transcriptomes of cultivated sweetpotato [Ipomoea batatas (L. ) Lam] and its wild ancestor [Ipomoea trifida(Kunth)G. Don ][J]. BMC Plant Biology, 2017, 17(1): 9.
[45]TAO X, GU Y H, JIANG Y Z, et al. Transcriptome analysis to identify putative floral-specific genes and flowering regulatory-related genes of sweet potato[J]. Bioscience, Biotechnology, and Biochemistry,2013, 77(11): 2169-2174.
[46]WEI C, LI M, QIN J, et al. Transcriptome analysis reveals the effects of grafting on sweetpotato scions during the full blooming stages[J]. Genes & Genomics, 2019, 41(8): 895-907.
[47]ALMOHANNA T, AHSAN N, BOKROS N T, et al. Proteomics and proteogenomics analysis of sweetpotato (Ipomoea batatas) leaf and root[J]. Journal of Proteome Research, 2019, 18(7): 2719-2734.
[48]楊倩春,李思宁,陈 硕,等. 代谢组学的运用及其研究进展[J]. 临床合理用药杂志, 2020, 13(2): 176-178.
[49]王佳钰. 重金属胁迫下植物代谢组学研究进展[J]. 绿色科技, 2020(1): 33-34.
[50]LEE W, YEO Y, OH S, et al. Compositional analyses of diverse phytochemicals and polar metabolites from different-colored potato (Solanum tubersum L.) tubers[J]. Food Science and Biotechnology, 2017, 26(5): 1379-1389.
[51]杨慧菊,兰玉倩,王石华. 植物响应低温胁迫组学研究进展[J]. 山东农业科学, 2020, 52(5): 142-148.
[52]DONADO-PESTANA C M, SALGADO J M, DE OLIVEIRA RIOS A, et al. Stability of carotenoids, total phenolics and in vitro antioxidant capacity in the thermal processing of orange-fleshed sweet potato (Ipomoea batatas Lam.) cultivars grown in Brazil[J]. Plant Foods for Human Nutrition, 2012, 67(3): 262-270.
[53]RAUTENBACH F, FABER M, LAURIE S, et al. Antioxidant capacity and antioxidant content in roots of 4 sweetpotato varieties[J]. Journal of Food Science, 2010, 75(5): C400-C405.
[54]KIM M Y, LEE B W, LEE H U, et al. Phenolic compounds and antioxidant activity in sweet potato after heat treatment[J]. Journal of the Science of Food and Agriculture, 2019, 99(15): 6833-6840.
[55]TEOW C C, TRUONG V, MCFEETERS R F, et al. Antioxidant activities, phenolic and β-carotene contents of sweet potato genotypes with varying flesh colours[J]. Food Chemistry, 2006, 103:829-838.
[56]ROSE I M, VASANTHAKAALAM H. Comparison of the Nutrient composition of four sweet potato varieties cultivated in Rwanda[J]. American Journal of Food and Nutrition, 2011, 1(1): 34-38.
[57]PARK S, LEE S Y, YANG J W, et al. Comparative analysis of phytochemicals and polar metabolites from colored sweet potato (Ipomoea batatas L.) tubers[J]. Food Science and Biotechnology, 2016, 25(1): 283-291.
[58]SU X, GRIFFIN J, XU J, et al. Identification and quantification of anthocyanins in purple-fleshed sweet potato leaves[J]. Heliyon, 2019, 5(6): e1964.
[59]WANG A, LI R, REN L, et al. A comparative metabolomics study of flavonoids in sweet potato with different flesh colors [Ipomoea batatas (L.) Lam ][J]. Food Chemistry, 2018, 260: 124-134.
[60]OKI T, MASUDA M, FURUTA S, et al. Involvement of anthocyanins and other phenolic compounds in radical-scavenging activity of purple-fleshed sweet potato cultivars[J]. Journal of Food Science, 2002, 67(5): 1752-1756.
[61]楊妍梅,李 玉,覃 圣,等. 静宁鸡PPARα基因克隆与生物信息学分析[J].江苏农业学报,2019,35(2):370-377.
[62]庞宁宁,樊怀福,王 哲,等. 黄瓜PP2-A1蛋白的生物信息学分析[J]. 江苏农业科学,2019,47(2):46-49.
[63]祖盘玉,李 维,林家栋,等. 赤水乌骨鸡TYR基因多态性及生物信息学分析[J].南方农业学报,2019,50(12):2806-2811.
[64]冯 磊,石元豹,汪贵斌,等. 银杏bHLH家族转录因子生物信息学及表达分析[J].江苏农业学报,2019,35(2):400-411.
(责任编辑:陈海霞)
