聚苯胺纳米线/SnO2复合光催化材料的光化学制备与性能
2021-06-29杨思娴钟文钰李超贤苏秋瑶许炳佳何谷平孙丰强
杨思娴,钟文钰,李超贤,苏秋瑶,许炳佳,何谷平,孙丰强,2
(1.华南师范大学化学学院,2.环境理论化学教育部重点实验室,广州 510006)
SnO2为n型氧化物半导体,其禁带宽度约为3.5 eV[1],与TiO2具有类似的光电化学性质且其原料易得、酸碱稳定性高,在光催化领域受到了广泛关注.但其自身光催化性能较差,因此,通过与其它材料复合促进其光生电子-空穴的分离,从而提高光催化活性成为了研究重点之一[2].迄今,国内外对SnO2基光催化复合材料已开展了一些研究,并获得了一系列复合材料,如采用水热结合煅烧的方法制备SnO2-ZnO 多孔片状复合材料[3],采用微波辐射法在离子液体中获得的SnO2-还原氧化石墨烯复合材料[4],静电纺丝结合高温煅烧法获得的TiO2/SnO2复合材料[5],二次光照法制备的聚噻吩/SnO2复合材料[6]等.显然,通过复合方式增强SnO2光催化性能已经成为一种有效手段,选择新材料、采用新方法进行有效复合并探讨光催化降解相应污染物是目前研究的总体趋势.SnO2与无机材料复合已有较多研究,但在复合时多涉及高温操作或复杂的前驱溶液设计.相对而言,SnO2与聚合物复合可通过室温方法实现,从绿色合成的角度具有突出的优势,同时可节省金属资源、降低催化剂成本,是光催化材料研究的趋势之一,但目前与SnO2复合的聚合物材料种类处于探索阶段,众多可增强SnO2光催化性能的聚合物尚待研究.
聚苯胺(PANI)是一种具有特殊电学、光学性质的共轭聚合物,经过掺杂后可具有优异的导电性及电化学性能[7],同时,其分子主链上含有大量的共轭π电子,当受适当波长光照射时,电子受激跃迁,出现附加的电子-空穴对[8].通过将PANI 与半导体复合可以在界面处形成异质结构,在适当波长光的激发下,相应光生电子和/或空穴将通过异质结快速转移,从而减少自身载流子的复合,提高半导体的光催化性能,因此,许多聚苯胺-半导体复合光催化材料被相继报道,并明显地增强了半导体的光催化性能,如PANI/TiO2[9],PANI/ZnO[10],PANI/AgFeO2[11]等.类似于其它半导体光催化材料,SnO2也与PANI具有匹配的能级结构,PANI同样具有增强SnO2光催化性能的潜力[12~14].但目前研究的PANI/SnO2复合材料均需要第三种成分辅助才能显示优良的增强效果,如柠檬酸辅助下增强复合材料光催化还原Cr(Ⅵ)离子[12],石墨烯[13]或多金属酸盐[14]增强复合材料降解污染物的性能,单纯的复合材料的光催化性能与PANI增强SnO2机制尚缺乏系统研究.同时,限于制备方法,目前复合材料的制备均是先制备SnO2纳米颗粒然后再复合PANI,且无论是SnO2还是PANI 均未进行形态的控制,易于导致3个不利于增强光催化性能的影响:(1)SnO2过多的活性位点被覆盖;(2)PANI属于聚合物材料,相对于无机材料其生长的择位性较弱,不易在SnO2纳米颗粒表面形成异质接触;(3)获得的复合材料呈无序分布的颗粒状态,易于造成颗粒的团聚.因此,从复合材料的制备方法入手,构筑高性能的PANI/SnO2复合材料并解决其中的性能增强机制,仍是促进SnO2在光催化领域应用的重要问题.
基于此,本文首先通过化学氧化法制备具有确定形态的PANI 纳米线,再将其分散在SnSO4和H2SO4的混合溶液中,利用光化学合成技术在室温下复合SnO2纳米颗粒,获得PANI纳米线/SnO2纳米颗粒复合材料,这可有效避免目前PAIN/SnO2复合材料制备的缺点,利于PANI增强SnO2的光催化性能.进一步以罗丹明B(RhB)染料为目标污染物,考查了不同反应条件制得的PANI/SnO2纳米线复合材料对RhB降解率的影响,发现PANI纳米线可以明显增强SnO2的光催化活性,并通过测试确定了复合材料的异质结构增强机制.
1 实验部分
1.1 试剂与仪器
苯胺(An,分析纯,广州化学试剂厂);过硫酸铵(APS,分析纯,天津致远化学试剂厂);硫酸亚锡(SnSO4,分析纯,上海麦克林生化科技有限公司);硫酸(H2SO4)、磷酸(H3PO4)和罗丹明B(RhB)均为分析纯(天津市大茂化学试剂厂).
BEJ-UV8W型紫外灯(佛山永轩达光电有限公司);752型紫外-可见分光光度计(UV-Vis,上海菁华科技仪器有限公司);UV-2700型紫外-可见漫反射光谱仪(UV-Vis DRS,日本岛津公司);D8 ADVANCE型X 射线衍射仪(XRD,德国Bruker 公司);K-Alpha 型X 射线光电子能谱仪(XPS,美国Thermo Fisher Scientific公司);Spectrum Two型傅里叶变换红外光谱仪(FTIR,德国Perkin Elmer公司);F-4600型荧光光谱仪(PL,日本日立公司);ZEISS Ultra 500 型扫描电子显微镜(SEM,德国Carl Zeiss 公司);JEM-2100HR型透射电子显微镜(TEM,日本JEOL公司).
1.2 实验过程
1.2.1 复合材料的制备 将0.20 mL苯胺放入烧杯中,滴加10 mL 0.10 mol/L磷酸与苯胺混合,在冰浴搅拌后加入0.20 g过硫酸铵继续搅拌至溶液呈墨绿色,将烧杯放入0 ℃低温环境反应24 h,取出离心并收集沉淀,将产物用去离子水反复洗涤至中性,在60 ℃下干燥6 h,得到PANI粉末.
称取0.50 g SnSO4溶解于100 mL 5.00 g/L H2SO4中,取0.05 g上述聚苯胺粉末加入其中,使聚苯胺充分分散,则前驱液中SnSO4/H2SO4/PANI 的质量比为1∶10∶0.1.随后将分散体系置于紫外灯下光照,调整紫外灯管高度控制液面的紫外光强度为0.70 mW/cm2,24 h 后离心分离收集沉淀,将沉淀用去离子水反复洗涤至中性,在60 ℃下干燥6 h,即得到光化学反应制备的SnO2/PANI复合材料.调整光照反应时间(3,6,15,24,36,48 h),相应制得的复合材料分别命名为SP3,SP6,SP15,SP24,SP36,SP48.
1.2.2 光催化降解实验 将0.05 g PANI/SnO2复合材料置于200 mL 玻璃烧杯中,加入100 mL 1.90×10-5mol/L的RhB溶液,在室温下超声均匀后置于暗处反应30 min后,取出5 mL反应样品,经离心过滤后取上层清液,在波长553 nm(此处对应于RhB的最大吸收系数)下测定原始吸光度A0,将取出的样品放回烧杯.再将其置于紫外灯(8 W,254 nm)下照射反应(紫外灯置于烧杯顶),每照射10 min取1次样品,经离心过滤后取上层清液,在波长553 nm下测定吸光度A.记录下各反应时间阶段的吸光度值,根据下式计算降解率,η(%):
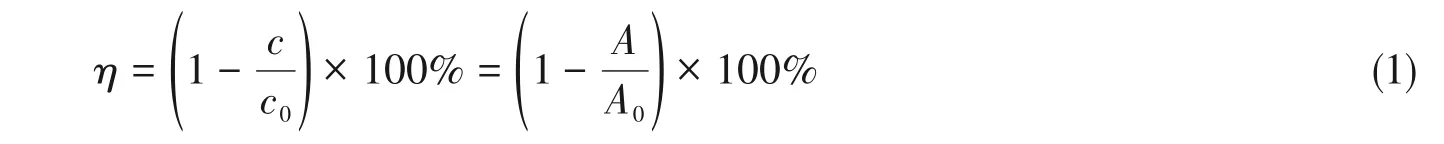
式中:c和c0(mol/L)分别为样品的降解后浓度和初始浓度;A和A0分别为样品光降解后和样品光辐照前在553 nm处的吸光度数值.
2 结果与讨论
2.1 复合材料的形态与成分表征
采用化学氧化法获得的纯PANI呈纳米线状结构[图1(A)],纳米线直径约为150~300 nm,长度超过3.5 μm,最大长径比在23以上.由光化学合成的SnO2呈颗粒状[图1(B)],颗粒近似呈球形、粒径不均匀、最大颗粒尺寸近700 nm,颗粒之间存在较多团聚.SnO2/PANI复合材料的形貌如图1(C)所示,球状的SnO2和纳米线状的PANI可清晰分辨,二者相互交织在一起,部分SnO2颗粒黏附在PANI线上,相比于纯的SnO2样品,SnO2颗粒因PANI线的阻隔而很少有团聚,进一步放大观察[图1(C)插图]发现部分SnO2纳米颗粒直接生长在PANI线上,这表明在紫外光照射下,SnO2纳米颗粒可很好地与PANI纳米线结合在一起形成复合物.图1(D)为SnO2,PANI和PANI/SnO2的XRD谱图.对比SnO2的XRD谱图和SnO2的标准卡片(PDF# 41-1445)发现,制备的SnO2为四方晶系金红石型结构,SnO2在2θ为26.6°,34.0°,51.7°处有明显的衍射峰,可对应于(110),(101)和(211)晶面.而PANI/SnO2相比SnO2衍射峰位置没有变化,说明聚苯胺的加入并没有改变SnO2的晶相结构.与SnO2相比,PANI/SnO2在2θ=51.7°处衍射峰的强度降低,这可能是PANI纳米线的引入影响了SnO2的生长,使SnO2纳米颗粒平均尺寸变小.合成的纯PANI 有一定的晶体特征,但在复合材料测试时,与PANI 对比,SnO2晶体衍射峰强度过高,导致PANI衍射峰难以显现.进一步利用透射电子显微对SnO2/PANI复合材料进行了观察,结果如图1(E)所示,PANI纳米线表面可以发现生长有SnO2纳米颗粒.选区1(Zone 1)的电子衍射[SAED,图1(E)插图]显示了SnO2的衍射环,说明形成的SnO2纳米颗粒呈多晶状态,通过衍射环的直径测量并结合XRD谱图可确定从内到外的衍射环可分别对应SnO2的(110),(101),(211)和(301)晶面衍射.无PANI的衍射信号说明PANI晶体特征不明显,更倾向于呈现非晶状态.选区1的高分辨透射电镜(HRTEM)照片[图1(F)]显示SnO2纳米颗粒(晶格条纹区)和PANI纳米线(无晶格条纹区)在界面处存在着原子尺度上的紧密接触,SnO2纳米颗粒由不同取向的晶粒或团簇组成,进一步显示了SnO2的多晶特征,晶面(101)和(110)的晶格条纹可清晰分辨,PANI纳米线因其非晶特点而无任何可分辨的晶格条纹.

Fig.1 SEM images of PANI(A),SnO2(B) and SnO2/PANI composite(C) and the corresponding XRD patterns(D),TEM(E)and HRTEM images(F)of SnO2/PANI composite
图2(A)为SnO2,PANI和PANI/SnO2复合材料的FTIR谱图.SnO2的谱线上,3200 cm-1左右的宽吸收带应归属为吸附水的伸缩振动;1622 和1495 cm-1处为物理吸附水中—OH 的变形振动吸收峰;1152,1045和960 cm-1处为SnO2表面不同类型羟基基团的弯曲振动峰[15];608 cm-1处的强吸收峰对应SnO2中Sn—O键的弯曲振动,为SnO2的特征吸收峰.在PANI红外光谱上[16],1591 cm-1处的吸收峰对应醌结构单元中C=N 键的伸缩振动;1509 cm-1处的吸收峰苯环结构中C=C 的伸缩振动;1300 cm-1处为C—N键的伸缩振动吸收峰;1048 和829 cm-1处的吸收峰分别对应苯环面内和面外C—H 键弯曲振动;620 cm-1处为苯环上的C—C 键的弯曲振动吸收峰.PANI/SnO2复合材料的红外光谱兼具SnO2和PANI 的典型特征吸收峰,但相比纯物质部分峰位出现了移动,如SnO2在608 cm-1处的吸收峰蓝移至612 cm-1处,PANI在829,1048和1509 cm-1处的吸收峰则分别红移至820,1043和1505 cm-1处,这说明PANI与SnO2之间存在强的相互作用,从而可实现紧密结合.进一步通过XPS 对复合材料进行了分析.图2(B)为PANI/SnO2复合材料的XPS全谱测试结果,除了C,N,O和Sn 4种元素不同原子轨道的光电子谱峰外,并未发现其它元素,说明复合材料在制备过程中未引入其它杂质.对其中的N1s峰分析见图2(C),从其拟合结果可以看出有3个分峰的存在,结合能为402.1 eV处的峰可对应于质子化亚胺(—N+=),结合能为400.2 与399.4 eV 的峰分别归属于苯二胺(—NH—)结构和醌二亚胺(—NH=)结构,具有质子化掺杂PANI 的特点[11].复合材料中的Sn3d谱峰存在2 个分峰[图2(D)],分别位于结合能为495.90和487.50 eV处,对应于Sn4+的Sn3d3/2和Sn3d5/2的结合能[17],但与纯SnO2结合能相比均提高了0.52 eV,这表明SnO2结合PANI 后影响了其电子能量分布[18],这进一步说明了PANI 与SnO2之间存在的强相互作用.

Fig.2 FTIR spectra(A) of PANI nanowires(a),PANI/SnO2 composite(b) and SnO2nanoparticles(c),XPS survey spectrum of PANI/SnO2(B),N1s XPS spectrum of composite(C)and Sn3d XPS spectra of composite and pure SnO2(D)
2.2 复合材料的形成机制
前驱液中,SnSO4主要作为锡源和光敏剂,H2SO4主要提供一个酸性环境,防止Sn2+水解.紫外光照射时,Sn2+将吸收光子形成Sn4+并释放电子[19,20],该电子在紫外光照射下以水合电子()的形态存在[21],属于强还原剂,可以直接还原Sn2+为金属Sn[E°(Sn2+/Sn0)=-0.14 V(vs.SHE)].新产生的Sn 团簇或纳米颗粒因具有较高的表面能而极易被溶液中的溶解氧氧化为SnO2.当然,在SnO2产生的同时,光生电子也会对H+进行还原产生H2.当未加入PANI时,SnO2将均匀成核并逐渐长大,并形成近似球形以尽可能降低表面能,同时,持续产生的H2具有类似搅拌的作用,对球形颗粒的产生以及减少颗粒间的团聚具有一定作用,反应过程如下所示:

除了上述由Sn2+引发的系列反应外,在光照下新生成的SnO2颗粒的光催化特性也将体现,即SnO2受光的激发后其价带电子可能跃迁到导带形成光生电子,如果导带能级合适,光生电子能会对Sn2+还原.同样道理,当将PANI纳米线分散在前驱溶液后也可能促进Sn2+的还原.为了确定SnO2和PANI的带隙(电子跃迁的可能性)以及导带能级,对光化学合成的SnO2纳米颗粒及PANI纳米线进行了UV-Vis DRS和价带XPS谱的测试与分析,如图3所示.SnO2对紫外光(波长200~380 nm)具有强吸收,PANI对200~800 nm 波长的光则均具有强的吸收.半导体的光学带隙可根据Kubelka-Munk 算法将DRS 进行转换[22]:

式中:α(L·mol-1·cm-1)为摩尔吸光系数;h为普朗克常数;v(Hz)为入射光频率;n值为2或1/2,依赖于半导体的类型及电子跃迁属性;B为比例常数;Eg(eV)为半导体的光学带隙.
SnO2和PANI均为直接带隙半导体,n值取2,则DRS转换的(αhv)2-hv曲线见图3(A)插图,分析可知Eg(SnO2)=3.26 eV,Eg(PANI)=2.06 eV.需要说明一点,对无机半导体来说其Eg为价带(VB)电子跃迁到导带(CB)所需的能量;对有机半导体则是其HOMO 轨道电子跃迁至LUMO 轨道所需能量,这里HOMO轨道和LUMO轨道类似于无机半导体的VB和CB.SnO2和PANI价带XPS谱见图3(B),从中可分析得到,SnO2和PANI的价带顶(VBM)分别为2.82和0.88 eV,此为样品和仪器的接触电势差值.根据经验的公式[23]:

式中:E(eV)(vs.NHE)为相对于一般氢电极的电势;Φ为仪器的功函数,本文为4.2 eV,VBMmeasured(eV)为直接根据价带XPS获得的价带顶能级.可获得SnO2的价带能级为2.58 eV(vs.NHE),导带能级为-0.68 eV(vs.NHE);PANI 的HOMO 和LUMO 轨道能级分别为0.64 和-1.42 eV(vs.NHE).由于254 nm紫外光的光子能量约为1240/254=4.88 eV,大于SnO2和PANI的带隙,在此光照下SnO2的VB电子和PANI的HOMO轨道电子将被激发到相应的CB和LUMO轨道,从而产生空穴和电子[反应式(8)和(9)].因为ECB(SnO2)和ELUMO(PANI)均比H+的还原电势[Eo(H+/H2)=0 eV]更负,所以SnO2的导带电子和PANI的LUMO轨道电子可以将H+还原为H2,同时,参考金属的活动顺序Sn2+同样可以被光生电子还原生成金属Sn[反应式(10)和(11)].通过该步骤产生的金属Sn也会进一步被氧化为SnO2.同时PANI纳米线具有良好的亲水性,其表面与溶液之间具有低界面能,根据晶体成核的一般理论,SnO2纳米颗粒可通过非均匀成核方式生长在纳米线上,从而可促成二者的紧密接触以及PANI/SnO2异质结构的产生.但由于SnO2颗粒形成速度过快,仍有很多颗粒通过均匀成核形成,最终获得SnO2纳米颗粒和PANI纳米线交织混合的形态,并有部分SnO2纳米颗粒直接生长在PANI纳米线上[见图1(C)和(E)].

Fig.3 UV-Vis DRS(A)and VB XPS spectra(B)of SnO2(a)and PANI(b)

2.3 复合材料的光催化性能
以RhB作为目标降解污染物,观察了不同光照时间获得的复合材料样品对RhB溶液在低功率紫外灯下(8 W,254 nm)的降解情况(图4).由图4(A)可见,纯聚苯胺几乎没有光催化活性,难以降解RhB;纯SnO2显示较好光催化活性,可使RhB在60 min内降解60.5%;复合材料SP3已显示一定光催化活性,可使RhB降解34.5%,但因复合材料中SnO2含量过少,与纯的SnO2相比活性仍较小;随着光照反应时间延长,SnO2含量逐渐增加,复合材料催化活性也逐渐提高,SP6的活性已高于纯SnO2,能够使RhB降解82.2%,SP15(曲线省略)和SP24则可使RhB分别降解91.5%和93.1%.进一步对RhB的光降解数据进行处理,发现所有材料参与的光降解反应均符合下面“准一级动力学”模型[24]:

式中:c0(mol/L)为RhB的初始浓度;c(mol/L)为降解反应时刻t时的RhB浓度;k(min-1)则为反应的表观反应速率常数.
如图4(B)所示,所有反应数据经过线性拟合均近似符合该一级线性方程,拟合系数(R2)均大于0.98,说明光降解反应非常满足这一动力学模型.这样k可反映材料的光催化活性,相关数据见图4(C),可明显看出复合材料制备时光照时间导致的复合材料活性变化规律与相对大小.随着光照时间增加,复合材料的光催化活性呈现先增大后减小的趋势,当光照时间超过6 h,可以获得比纯SnO2活性高的复合材料样品,其中光照24 h的样品具有最高活性,是纯SnO2的2.8倍多.显然,通过光化学方法使SnO2与PANI纳米线复合是提高和调控SnO2光催化活性的一种有效手段.

Fig.4 Photocatalytic degradation of RhB by composite,pure PANI and SnO2
2.4 复合材料光催化性能的增强机制
水溶液中的光催化降解反应一般过程为:半导体材料受光照产生光生电子和光生空穴,光生电子进一步与溶解氧反应生成超氧自由基(·),·在H+或H2O的参与下也可转化为羟基自由基,光生空穴在在H+或H2O的参与下转化为·OH,强氧化性的·OH,·或空穴使污染物氧化分解.

对光降解反应起作用的氧化剂物种,取决于光催化材料和污染物种类,可通过在光催化实验中加入相应捕获剂来确定.图5(A)示出了SP24 光催化降解RhB 过程中分别通入N2气(阻止·O2-的产生)、滴加甲醇(Methanol,捕获空穴)、滴加叔丁醇(tert-Butanol,捕获·OH)以及滴加叔丁醇的同时通入N2(捕获·OH并阻止·的产生)时的对比情况.显然,阻止某一氧化剂物种的产生均可导致光催化降解率的下降,说明在此光催化降解反应中有·、·OH与空穴的产生并均对RhB有氧化分解作用.滴加叔丁醇并同时通入N2气与单纯通入N2气或单纯滴加叔丁醇时相比,RhB 的降解率明显减低得更多,说明·OH的产生与·O2-和空穴均有关系,即光催化反应过程中存在e-→·→·OH和h+→·OH 2条转化途径.即载流子(e-和h+)的数量将会影响氧化剂的浓度,进而影响光催化效率.通常,光催化过程中同时伴随着光生载流子之间的复合,因此,减少载流子复合提高其分离效率是增强光催化性能的重要手段.鉴于此,对SP24,SnO2与PANI的光致荧光发射进行了测试和分析,结果如图5(B)所示.在约426 nm处三者均存在的荧光发生峰,但复合材料的荧光发射强度明显低于单一物质,说明光生载流子在复合材料中具有更高的分离效率,从而可更有效地被利用,提高光催化活性.
通过对以上数据的分析可知,PANI 增强SnO2光催化活性应归因于它们之间形成的异质结构.从前述测试分析(图3)可知,PANI 的LUMO 和HOMO 轨道能级分别为-1.42 和0.64 eV(vs.NHE),SnO2的CB 和VB 能级分别-0.68 和2.58 eV(vs.NHE),单纯从能级结构看似乎二者可形成Ⅱ型异质结(Type Ⅱheterojuction),价带与导带能级交错排列,PANI的LUMO轨道能级比SnO2的CB能级更负,电子可向SnO2转移,SnO2的价带空穴向PANI 的HOMO 轨道转移.因为PANI 的HOMO 轨道能级比OH-/·OH的氧化还原电势(2.43 eVvs.NHE)[25]更负,光催化反应中由h+→·OH的反应将不可能发生,但光催化反应时的·OH捕获实验[图5(A)]结果已证实部分·OH由h+转化而来,这与该假设存在矛盾.事实上,决定电子转移的因素应该为二者的费米能级差异,即电子从高费米能级的物质向低费米能级物质转移.PANI 为p 型半导体,其功函数在4.6~4.7 eV 之间[26],SnO2为n 型半导体,其功函数为4.3~4.5 eV[27],这意味着SnO2的费米能级要高于PANI 的费米能级,SnO2的电子应向PANI 转移,二者组成的异质结应当为Z 型异质结(z-Scheme heterojuction)[28],界面处形成一个内部电场,方向为SnO2→PANI.SnO2的CB能级比PANI的HOMO轨道能级更负,从而有利于SnO2的CB电子通过异质结进入PANI的HOMO轨道,即异质结可视为一个电子快速通过的通道.基于此,SnO2/PANI复合材料光催化降解RhB的反应机制如图6所示.
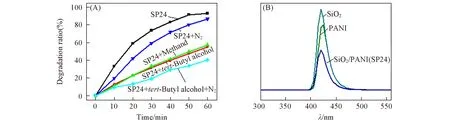
Fig.5 Degradation of RhB after the addition of capture agent in photocatalytic reaction(A) and photoluminescence spectra of SP24,SnO2 and PANI(B)
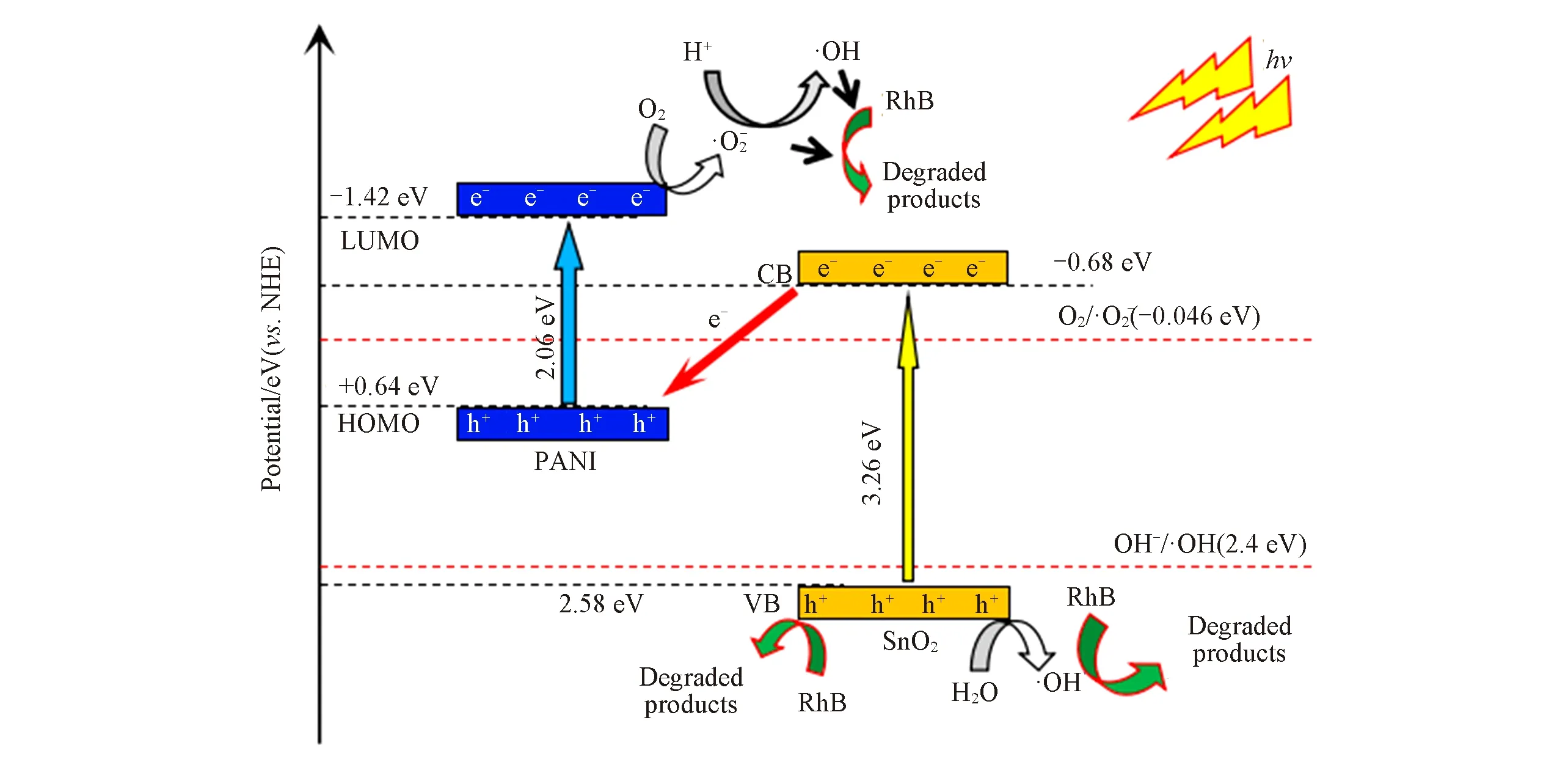
Fig.6 Schematic diagram of photocatalytic degradation reaction mechanism of RhB caused by PANI/SnO2 composites
在RhB溶液中当复合材料受到紫外光照射时,SnO2的VB电子被激发到CB形成光生电子[式(8)],同时,PANI的HOMO轨道电子也会激发到LUMO轨道[式(9)],在相应的VB和HOMO轨道上会留下空穴(h+).然后,SnO2的导带电子将迅速通过异质结进入PANI的HOMO轨道与空穴复合,这样可有效阻止SnO2的VB空穴被光生电子复合以及阻止PANI的LUMO轨道电子与空穴复合,从而更多的载流子可转化为强氧化性自由基或直接被利用.SnO2的价带能级比(H2O或OH-)/·OH的氧化还原电势更正[28],其光生空穴可将OH-或H2O转化为羟基自由基(·OH)[式(13)];PANI的LUMO轨道能级比O2/·的氧化还原电势(-0.046 eVvs.NHE)更负,其光生电子可将表面吸附的O2转化为超氧自由基(·)[式(14)],进一步,部分·可转化为·OH[式(15)].生成的·OH和·以及SnO2的价带空穴均具有强氧化性,均可氧化分解RhB 分子并最终使其转化为CO2、H2O 及其它无机物从而达到处理污染物的目的[式(16)][29,30].显然,SnO2与PANI的复合因异质结的形成促进了相应光生电子与空穴的有效分离,进而实现了光催化活性的提高,相反,纯物质由于电子与空穴易于复合而不能被有效利用[图5(B)],从而具有较低光催化活性.
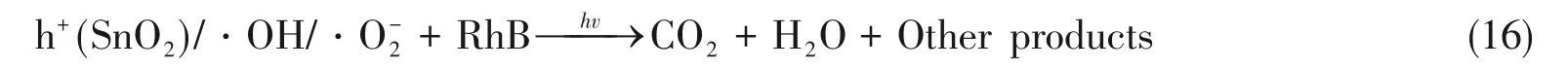
当然,增强光催化活性的效果与SnO2的生成量也密切相关,当SnO2生成时间过短(如3 h),SnO2生成量少,形成异质结的数量少,在相同光催化剂质量的情况下,在光催化反应时与纯SnO2相比所产生的光生载流子过少,尽管有PANI的增强作用,但其活性仍然比纯SnO2低;随着生成时间增加,SnO2在复合材料中的含量增加,异质结数量增多,产生的载流子数量增加,PANI增强效果提高,从而使复合材料的活性逐渐增加[31,32];生成时间为6 h时复合材料的活性已大于纯SnO2,控制生成时间为24 h时,所获得复合材料在光催化反应时载流子产生的数量与异质结数量达到最佳匹配,具有最高的光催化活性;生成时间继续增加(如36和48 h),则SnO2生成量过多,将覆盖过多的PANI纳米线表面,在光催化反应时将影响PANI纳米线的受光面积及其与O2的接触,影响由PANI产生的光生载流子数量及·的产生,从而使其活性逐渐降低,但因较多异质结的存在,由SnO2产生的载流子仍然可以有效分离,其活性仍然远大于纯SnO2,这进一步说明异质结的形成是PANI增强SnO2光催化活性的主要原因.综合以上实验结果,SnO2的光照生成时间24 h具有最佳光催化活性.需要指出的是,这个优选时间是基于前驱液中各物质的特定浓度比确定的,不难理解,当比值变化时,相应的优选复合时间需要通过进一步的实验获得.
3 结 论
以PANI 纳米线为基础,采用室温光化学方法实现了PAIN 纳米线/SnO2新型复合光催化材料的制备,其光催化性能与纯SnO2和纯PANI相比均具有明显提高,且随着光照生成SnO2时间的延长,所获得的复合材料在降解RhB时光催化活性呈现先提高后降低的规律,在最优光照生成时间下得到复合材料的光催化活性是纯SnO2的近3倍.复合材料性能增强的原因在于二者形成的Z型异质结促进了SnO2导带电子向PANI的HUMO轨道转移,减少了SnO2价带空穴和PANI的LUMO轨道电子的损失,进而促进了·OH和·O2-活性物种的生成.以具有确定形态的聚合物为基础进行生长复合,对于SnO2基复合材料来说是一种新的复合方式,将使复合材料的性能易于调控和优选,同时,光化学方法具有绿色、廉价、易于操作的特点,这将为构筑半导体/聚合物高性能光催化复合材料提供一种新的途径,并促进该类材料在环境污染物处理以及光解水产氢方面的应用.
