掌叶大黄“柴禾”熏制加工过程中成分及体外抗氧化活性动态变化
2021-06-26冯银平帖晓燕张云鹤张文广苗小楼李秀娟
冯银平,帖晓燕,张云鹤,王 丹,张文广,苗小楼,李秀娟,李 芸*
(1.甘肃中医药大学,甘肃 兰州 730000;2.中国农业科学院兰州畜牧与兽药研究所,甘肃 兰州 730050;3.甘肃省中医院,甘肃 兰州 730050)
掌叶大黄Rheum palmatum L.是《中国药典》收载的正品大黄之一[1],主要含有蒽醌类、蒽酮类、二苯乙烯类、苯丁酮类、色原酮类、黄酮类、鞣质类、多糖类等成分[2],其中蒽醌类是其重要的活性物质,也是现行版《中国药典》 规定控制大黄药材质量的指标,具有抗炎、抗肿瘤、保护心血管、保肝、护肺、改善脑损伤、调节肠道菌群、治疗肾纤维化等生物活性[3-5]。
甘肃礼县是掌叶大黄的传统道地产区,种植历史悠久,但鲜品含水量较高,根茎粗大,质地坚实,水分不易散失,又因礼县气候潮湿阴冷,采收期处于秋冬季,易导致药材霉烂、糠心、变色、变质,故采收后及时的干燥处理方式对保证药材质量和饮片临床疗效至关重要。大黄传统干燥方法主要有熏干法、烘干法、阴干法等[6],但烘干法对设备要求高导致生产成本高,而阴干法历时较长,易虫蛀霉变,因而当地药农普遍采用符合农家生产加工模式的熏干法。历代本草对大黄药材干燥方法均有记载[7-9],宋平顺等[10]发现,柴禾熏制可使掌叶大黄蒽醌类成分含量升高,但关于柴禾熏制加工过程中化学成分和药效的变化情况至今尚无报道。因此,本研究从以上2 个方面出发,探讨柴禾熏制不同阶段掌叶大黄成分及体外抗氧化活性的动态变化规律,初步揭示药材产地熏制加工机理。
1 材料
1.1 仪器与试剂 Agilent 1260Ⅱ型高效液相色谱仪、Agilent Extend-C18色谱柱(4.6 mm×250 mm,5 μm)(美国Agilent 公司);BT125D 型电子天平、MA 37 型快速水分测定仪(德国赛多利斯公司);Milli-Q 超纯水系统(德国默克生命科学公司);FW-400A 型高建万能粉碎机(北京科伟永兴仪器有限公司);DK-98-Ⅱ型电热恒温水浴锅(天津市泰斯特仪器有限公司);R-200 旋转蒸发仪(瑞士Buchi 公司);SHB-Ⅲ循环水式多用真空泵(郑州长城科工贸有限公司);DHG-9240A 型电热恒温鼓风干燥箱(上海一恒科技有限公司);KQ-300VDE型超声波清洗器(昆山市超声仪器有限公司);Multiskan GO 多功能酶标仪(美国赛默飞世尔科技有限公司);96 孔细胞培养板(北京鼎国生物技术有限公司)。
芦荟大黄素(批号CHB160628)、大黄素甲醚(批号CHB150817)、大黄酸(批号CHB160628)、大黄素(批号 CHB150527)、大黄酚(批号CHB160914)对照品均购自成都克洛玛生物科技有限公司,纯度≥98%。色谱纯甲醇、磷酸(天津市大茂化学试剂厂);1,1-二苯基-2-三硝基苯肼(DPPH)(美国Sigma-Aldrich 公司);维生素C(中国医药工业公司北京制药厂);其他试剂均为分析纯;水为甘肃中医药大学科研实验中心分析测试一实验室自制超纯水。
1.2 样品 掌叶大黄药材采自甘肃礼县春天药业有限公司同一种植基地,委托当地药农产地柴禾熏制加工,经甘肃中医药大学附属医院杨锡仓主任中药师鉴定为蓼科植物掌叶大黄Rheum palmatum L.的根及根茎。
2 方法与结果
2.1 熏制品制备 新鲜掌叶大黄采收后去掉支根,从中间切成两半,置于屋内棚上,使其切口向下,在棚下炉中点燃柴禾,不用明火,而用其烟熏制,直至完全熏干。根据熏制天数不同,分2 年共12批样品,分别记为S1.1~S1.7(熏制0、10、15、30、40、60、120 d)及S2.1~S2.5(熏制0、60、100、140、180 d),其中样品S2.5 为熏制140 d 后下架自然晾干而来。色谱图见图1。
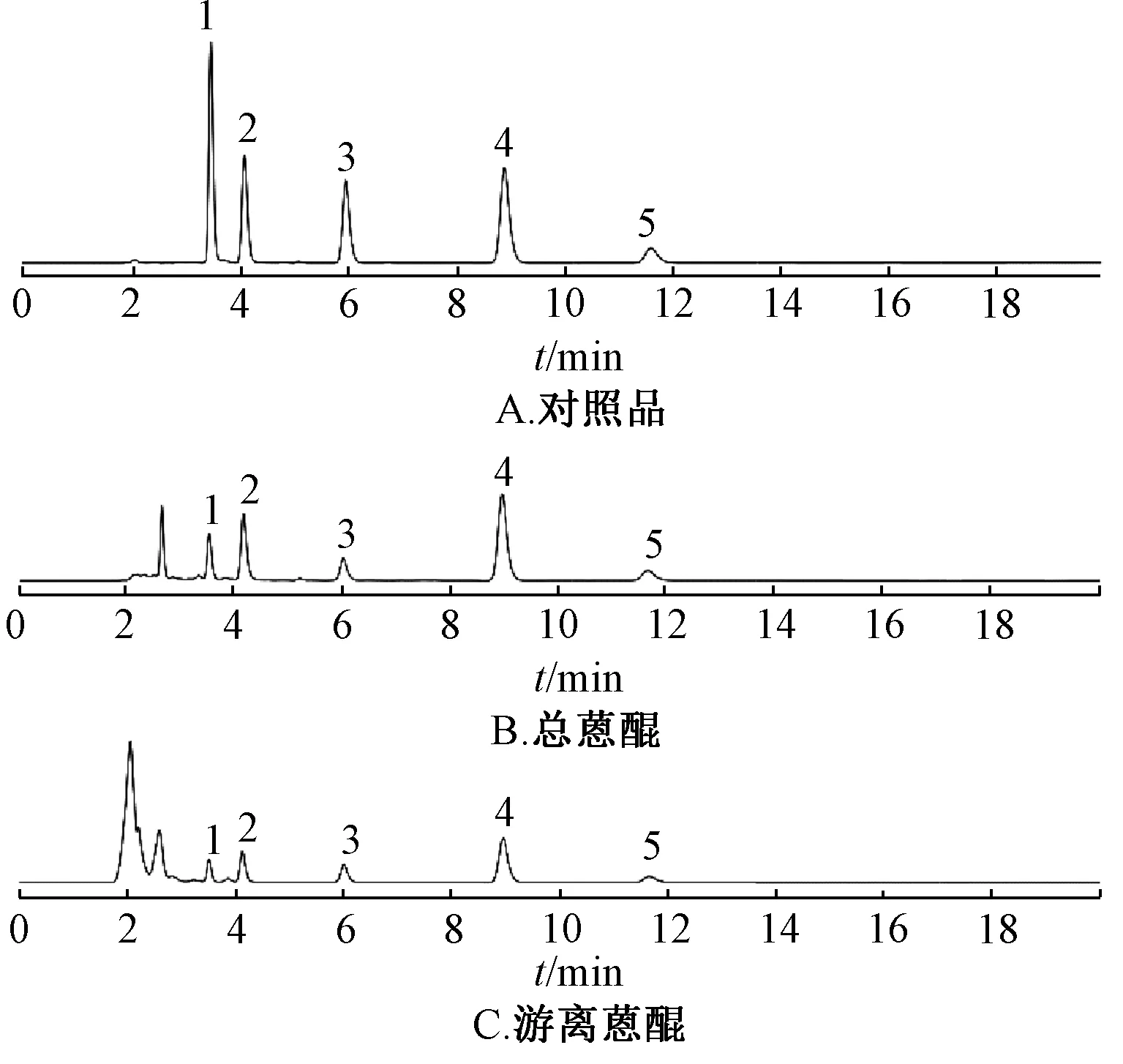
图1 各成分HPLC 色谱图Fig.1 HPLC chromatograms of various constituents
2.2 水分、浸出物测定
2.2.1 水分 将样品粉碎,混合均匀,四分法取样,采用快速水分测定仪测定水分,平行3 次,取平均值,结果见图2。
2.2.2 浸出物 取样品(按干燥品计)约2 g,精密称定,按2015 年版《中国药典》 四部(通则2201)水溶性浸出物测定项下热浸法进行浸出物测定,结果见图2。
2.3 总蒽醌、游离蒽醌含量测定
2.3.1 色谱条件 参考文献[1],Agilent Extend-C18色谱柱(4.6 mm×250 mm,5 μm);流动相甲醇-0.1%磷酸(85∶15);体积流量1 mL/min;柱温40 ℃;检测波长254 nm;进样量10 μL。
2.3.2 对照品溶液制备 精密称取芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚对照品适量,加甲醇分别制备成质量浓度为84、86、78、84、40 μg/mL 的溶液,各精密吸取2 mL,混匀,即得。
2.3.3 总蒽醌供试品溶液制备 取本品粉末(过4 号筛)约0.15 g,精密称定,参考文献[1] 报道的方法处理,即得。
2.3.4 游离蒽醌供试品溶液制备 取本品粉末(过4 号筛)约0.5 g,精密称定,参考文献[1]报道的方法处理,即得。
2.3.5 样品含量测定 取“2.3.2”下对照品溶液与“2.3.3”“2.3.4”下供试品溶液,在“2.3.1”项条件下测定,计算含量,色谱图见图1,结果见图2。
2.4 DPPH 自由基清除率测定
2.4.1 DPPH 贮备液制备 精密称取DPPH 试剂2.0 mg,甲醇溶解定容至25 mL,即得(80 μg/mL),避光置于4 ℃冰箱中冷藏备用。
2.4.2 样品贮备液制备 取各样品粉末约0.5 g,精密称定,精密加入60% 乙醇15 mL,超声(300 W)提取30 min,抽滤,滤液旋干,残渣加甲醇溶解[11],定容至100 mL,取1 mL,甲醇定容至10 mL,即得(500 μg/mL),放入4 ℃冰箱中冷藏,以配制不同质量浓度的样品溶液。
2.4.3 维生素C 对照液制备 取维生素C 适量,加甲醇溶解,制备成质量浓度为100 μg/mL 的溶液,即得。
2.4.4 自由基清除率测定 取上述溶液,分别稀释成25、50、100、200、300、400、500 μg/mL 和5、10、20、40、60、80、100 μg/mL,在96 孔板上加入不同质量浓度的样品、DPPH 溶液各100 μL,室温避光反应30 min,于517 nm 波长处测定吸光度Ai、100 μL 甲醇与100 μL DPPH 溶液混合后吸光度Ao、100 μL 样品溶液与100 μL 甲醇混合后吸光度Aj,平行3 次,取平均值,按公式[1-(Ai-Aj)/Ao] 计算清除率,同法处理维生素C 对照液,结果见图3。
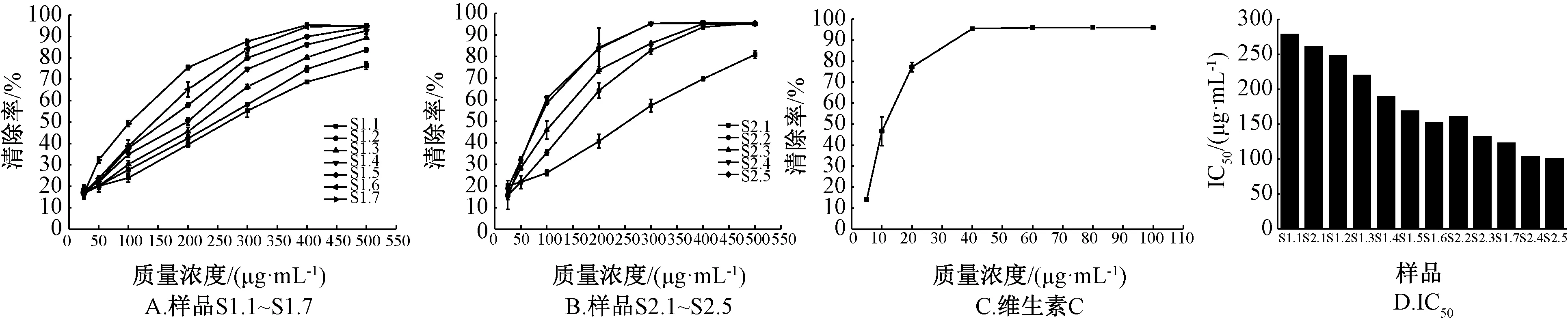
图3 各样品DPPH 自由基清除率与IC50值Fig.3 DPPH free radical scavenging rates and IC50 values of various samples
2.4.5 IC50值计算运用SPSS 19.0 软件中的Probit 分析功能,以清除率为响应频率,观测值汇总100%,质量浓度为协变量,模型为Logit。
2.5 相关性分析
2.5.1 熏制天数与各指标 以熏制天数为自变量,水分、浸出物、总蒽醌含量及抗氧化活性(IC50)为因变量进行Pearson 相关性检验。结果显示,熏制天数与水分、总蒽醌、IC50的相关系数分别为-0.906(P=0.000)、0.888(P=0.000)、-0.920(P=0.000),双侧P 均<0.01,提示熏制天数与水分呈负直线相关,与总蒽醌含量和抗氧化活性呈正直线相关;与浸出物的相关系数为0.631(P=0.028),双侧P<0.05,结合图2 可认为熏制天数与浸出物整体呈正相关,随着熏制天数的增加浸出物先呈上升趋势,在60 d 后逐渐趋于稳定。
2.5.2 抗氧化活性(IC50)与各指标 以抗氧化活性(IC50)为自变量,水分、浸出物、总蒽醌含量为因变量进行Pearson 相关性检验。结果显示,IC50与水分、浸出物、总蒽醌的相关系数分别为0.961(P=0.000)、-0.751(P=0.005)、-0.766(P=0.004),双侧P 均<0.01,提示抗氧化活性与水分呈负相关,与浸出物、总蒽醌含量呈正相关。
2.5.3 其他指标 水分与浸出物的相关系数为-0.753(P=0.005),与总蒽醌的相关系数为-0.811(P=0.001),双侧P 均<0.01,表明水分与浸出物、总蒽醌含量呈负相关。
游离蒽醌含量与各项指标之间均无相关性,结合t 检验可知,随着熏制天数增加,两相邻采样点游离蒽醌含量变化显著(P<0.05),整体呈近“M”型变化趋势,表明熏制过程中游离蒽醌变化复杂,但机制尚不明确。
2.6 化学模式识别
2.6.1 主成分分析 以12 批熏制不同天数样品的水分、浸出物、总蒽醌、游离蒽醌、IC50值作为特征变量值,将数据导入SIMCA 14.1 软件进行主成分分析,以主成分得分及各变量载荷为依据,结果见图4。由此可知,第1 主成分的方差百分比为65.364%,第2 主成分的方差百分比为19.875%,累积方差百分比为85.239%,可以表征原始数据特征;12 批样品聚为4 大类,从左至右分别为熏制120~140 d、熏制40~100 d、熏制15~30 d、熏制0 d,对应药材质量为“最优”“较好”“次之”“较差”,表明熏制不同天数药材所含水分、浸出物、总蒽醌、游离蒽醌含量及抗氧化活性存在显著差异,所建立的模型良好,可以对不同质量药材进行有效区分。由图5 可知,水分、浸出物、总蒽醌及IC50在第1 主成分中具有较大载荷,表明熏制天数影响水分、浸出物、总蒽醌含量及抗氧化活性,四者之间可能存在交互作用;第2 主成分与游离蒽醌密切相关,表明掌叶大黄药材经熏制后游离蒽醌变化明显。
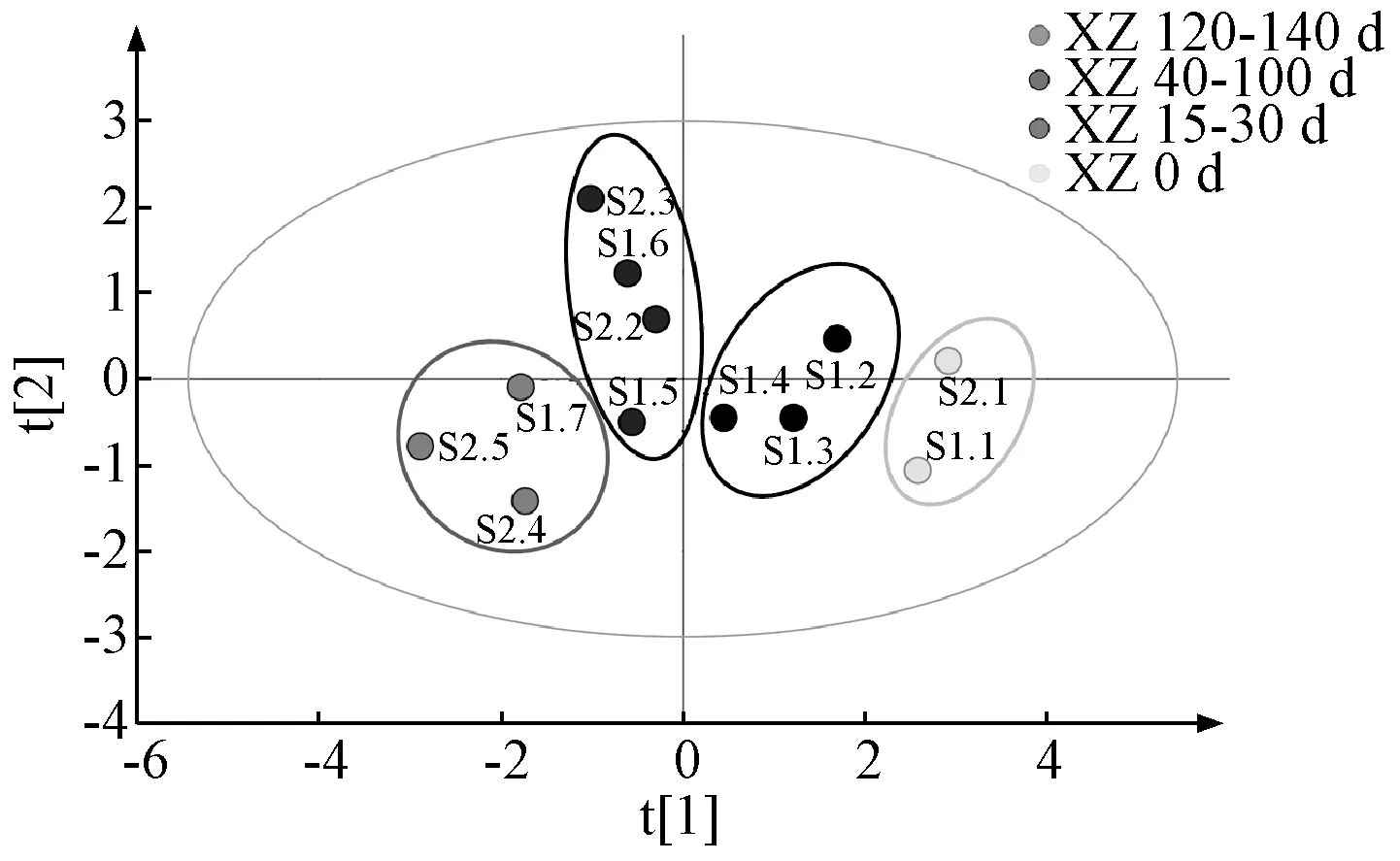
图4 12 批样品主成分分析图Fig.4 Principal component analysis plot for twelve batches of samples
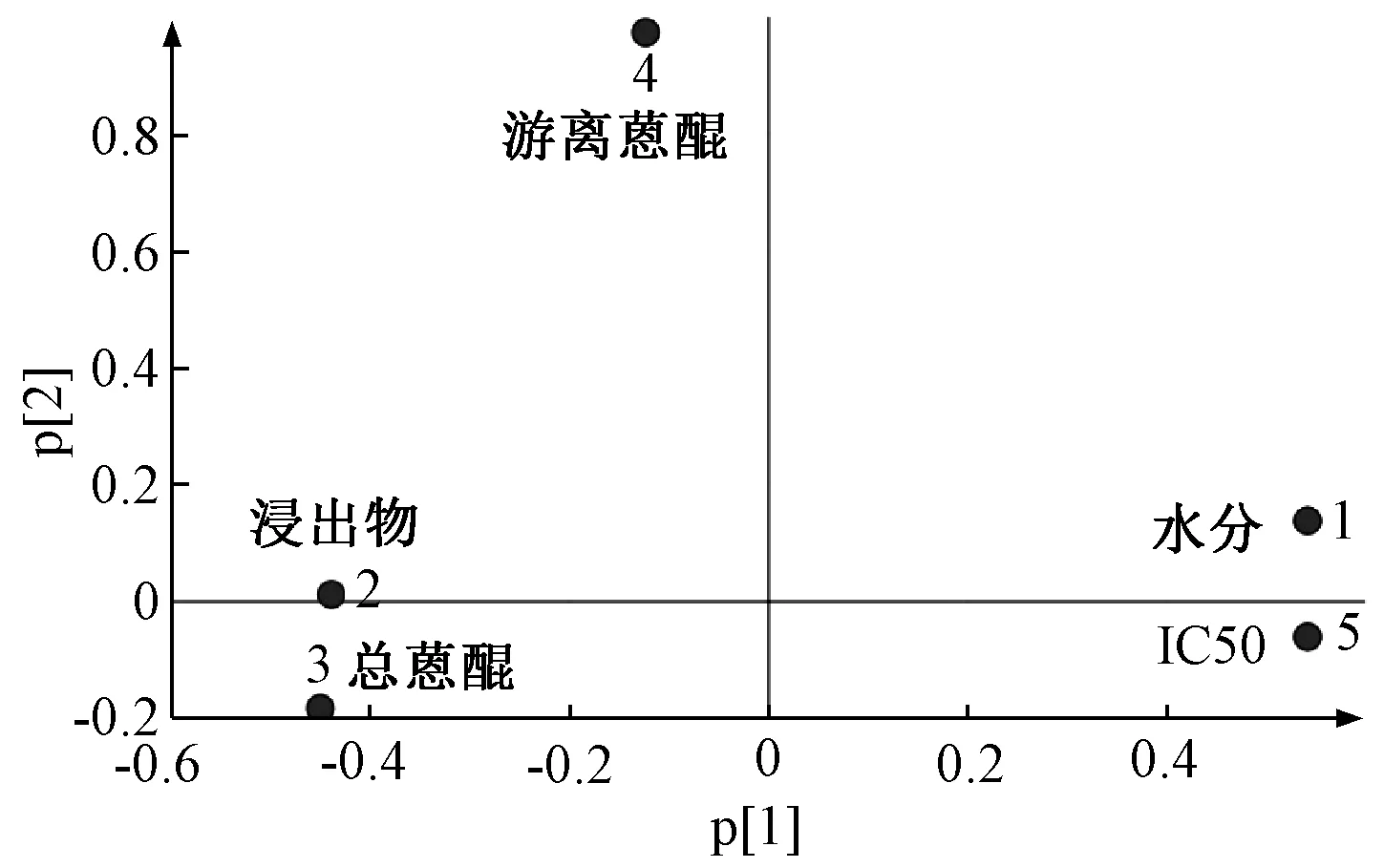
图5 12 批样品各变量载荷图Fig.5 Variable load graph of twelve batches of samples
2.6.2 偏最小二乘判别分析(PLS-DA)在主成分分析的基础上进行PLS-DA[12],获得样品分布散点图,见图6。由此可知,12 批样品根据熏制天数被分为4 类,表明不同熏制天数下药材差异显著。
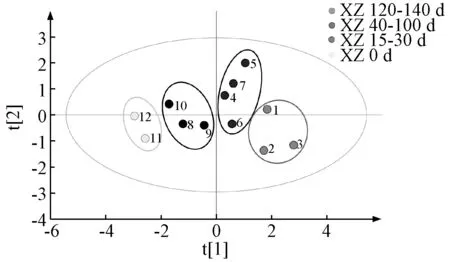
图6 各样品OPLS-DA 得分散点图Fig.6 OPLS-DA score scatter plot for various samples
12 批样品PLS-DA 模型中各变量的VIP[13]见图7,VIP 越大,表明该变量对于样品分类的贡献越大。由此可知,5 个变量对于差异性的影响程度依次为游离蒽醌>IC50>水分>总蒽醌>浸出物,均为重要变量(VIP>0.5),其中游离蒽醌的VIP>1.0,表明其变化是导致药材质量差异的重要因素。
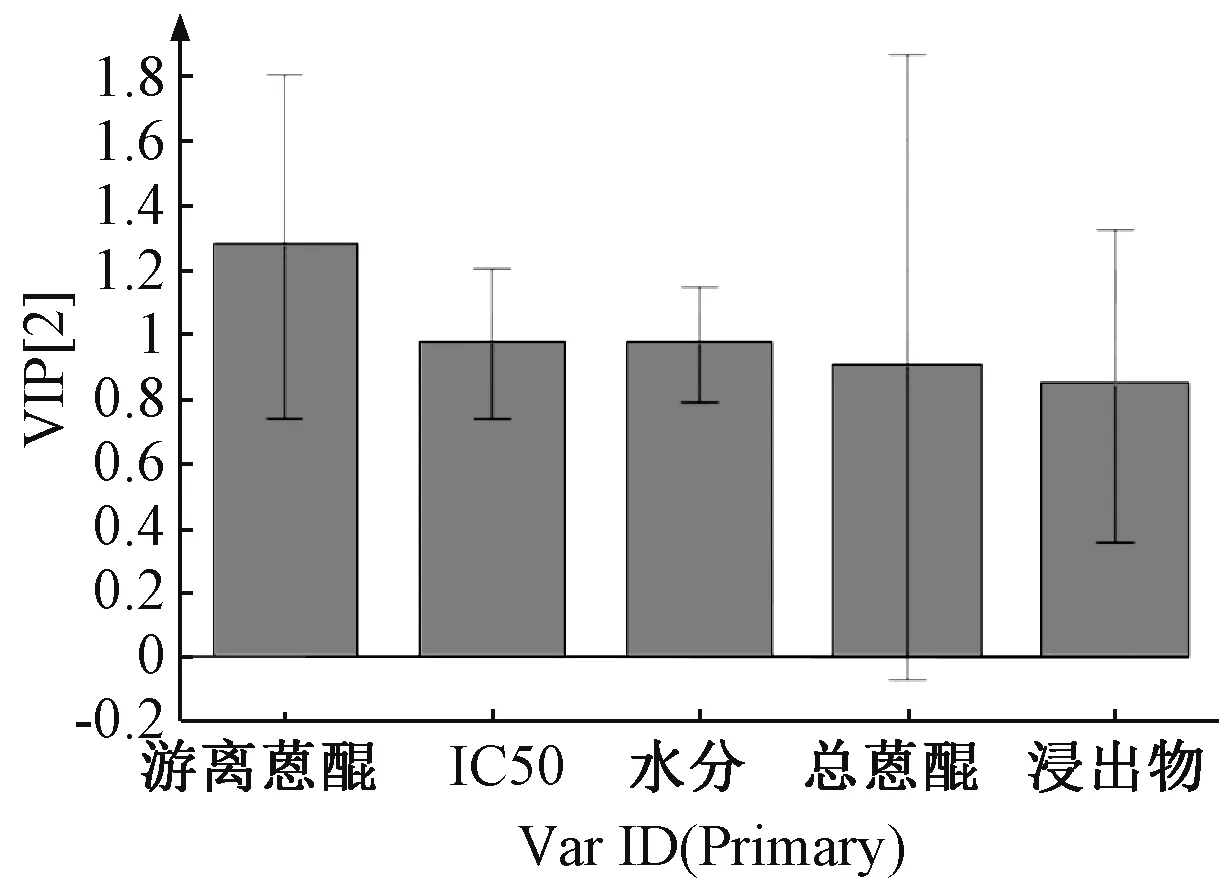
图7 各样品PLS-DA VIP 图Fig.7 PLS-DA VIP plot of various samples
3 讨论
自由基是单质或化合物均裂产生的带有未成对电子的原子或基团,研究发现其与心血管疾病(CVD)、恶性肿瘤、2 型糖尿病、感染机制、纤维形成和神经紊乱等多种疾病的发生有密切的关系[14-15]。研究发现,大黄提取物的抗氧化活性与蒽醌类成分有一定的相关性[16]。课题组前期以大黄加工品的折干率、蒽醌衍生物含量及横切面质地和色泽变化为指标,比较了晒干法、阴干法、微波法、不同温度烘干和传统柴火熏制等不同干燥方式,结果显示不同干燥方式以熏干法的蒽醌类成分含量、横切面色泽质量最好[17]。
熏干法的具体操作参考文献[18],本研究结果表明,掌叶大黄药材在熏制过程中化学成分和体外抗氧化活性发生显著变化。随着熏制天数增加,水分降至安全范围,浸出物和抗氧化活性先增加后趋于稳定,总蒽醌含量呈“单项递增”模式,游离蒽醌“先增后减,再增再减”,即近“M”型变化模式。蒽醌类成分所呈现的变化模式在其饮片炮制过程中也有类似表现,杨丽等[19]在研究大黄炭加热过程颜色特征与14 种成分含量变化关系时发现随着加热温度升高,游离蒽醌呈“先增后减”变化趋势,并推测导致该变化的原因可能是温度升高导致结合蒽醌苷键断裂形成游离蒽醌,而加热时间过长后又使得游离蒽醌结构被破坏,游离蒽醌的羰基先降解由苯醌转化成萘醌,再进一步转化成醛或酮;另外有关大黄发酵使结合蒽醌转化为游离蒽醌的研究报道较为常见[20],而综合炒炭、发酵等炮制方法发现均与发热现象或微生物的参与有关,推测大黄药材采收之后,在一定含水量、适宜温度、相关微生物酶的参与下,其次生代谢产物蒽醌类成分的转化和积累仍十分活跃,故总蒽醌含量呈“单项递增”模式,而游离蒽醌的变化较为复杂。课题组将在后期进行相关微生物酶方面的研究,以期明确其变化机理,为进一步阐明熏制加工机制和规范产地加工技术提供参考。
致谢:衷心感谢甘肃礼县春天药业有限公司、兰州大学胡芳弟教授、甘肃省中医院闫治攀主管中药师以及课题组成员马冬妮、杨秀娟、石琪奇等对本研究提供的指导和帮助。
