Genome-wide identification,molecular evolution,and expression divergence of the hexokinase gene family in apple
2021-06-24ZHULingchengSUJingJINYuruZHAOHaiyanTIANXiaochengZHANGChenMAFengwangLIMingjunMABaiquan
ZHU Ling-cheng,SU Jing,JIN Yu-ru,ZHAO Hai-yan,TIAN Xiao-cheng,ZHANG Chen,MA Fengwang,LI Ming-jun,MA Bai-quan
1 State Key Laboratory of Crop Stress Biology for Arid Areas/Shaanxi Key Laboratory of Apple,College of Horticulture,Northwest A&F University,Yangling 712100,P.R.China
2 Department of Plant Biology,University of Illinois at Urbana-Champaign,Urbana,IL 61801,USA
Abstract Hexokinase (HXK) is the first irreversible catalytic enzyme in the glycolytic pathway,which not only provides energy for plant growth and development but also serves as a signaling molecule in response to environmental changes. However,the evolutionary pattern of the HXK gene family in apple remains unknown. In this study,a total of nine HXK genes were identified in the Malus×domestica genome GDDH13 v1.1. The physiological and biochemical properties,exonintron structures,conserved motifs,and cis-elements of the MdHXK genes were determined. Predicted subcellular localization indicated that the MdHXK genes were mainly distributed in the mitochondria,cytoplasm,and nucleus. Gene duplication revealed that whole-genome duplication (WGD) and segmental duplication played vital roles in MdHXK gene family expansion. The ω values of pairwise MdHXK genes indicated that this family was subjected to strong purifying selection during apple domestication. Additionally,five subfamilies were classified,and recent/old duplication events were identified based on phylogenetic tree analysis. Different evolutionary rates were estimated among the various HXK subfamilies. Moreover,divergent expression patterns of the MdHXK genes in four source-sink tissues and at five different apple fruit developmental stages indicated that they play vital roles in apple fruit development and sugar accumulation. Our study provides a theoretical basis for future elucidation of the biological functions of the MdHXK genes during apple fruit development.
Keywords:apple,hexokinase,cis-element screening,evolutionary pattern,sugar accumulation
1.Introduction
In higher plants,sugars play many important roles,not only as energy sources and structural components for growth and development but also as central signaling molecules that control the expression of genes involved in many vital processes,such as photosynthesis,senescence,growth,pathogen defense,and biotic and abiotic stress responses (Ruan 2014;Zhuet al.2021).Sucrose,the main photosynthetic product,is synthesized in source leaves and then translocated to nonphotosynthetic sink organs through the phloem (Patricket al.2013). In sink organs,sucrose is either hydrolyzed into glucose (Glc) and fructose (Fru) by invertase or cleaved into UDP-Glc and fructose by sucrose synthase (Wanet al.2017). The resulting hexoses (glucose and fructose) are then phosphorylated by hexokinase (HXK) and fructokinase (FRK),and the phosphorylated hexoses participate in various plant metabolic processes (Granot 2007).
HXK is an essential enzyme that catalyzes the phosphorylation of hexose into hexose-6-phosphate during the first step of the glycolytic pathway,and HXK has been identified in many plant species,such asArabidopsis,rice,maize,cassava,and pear (Choet al.2006;Karveet al.2008;Zhanget al.2014;Genget al.2017;Yuet al.2018). InArabidopsis,AtHXKgenes play dual functions as catalysts and hexose sensors by integrating nutrient,light,and hormone signaling (Janget al.1997;Mooreet al.2003). Among these genes,AtHXK1-3can phosphorylate hexose,while the other three members,AtHKL1-3,lack this catalytic activity and only mediate sugar sensing (Karveet al.2008). In addition,HXKsregulate the gene expression involved in sucrose metabolism and starch biosynthesis.VvHXK1andVvHXK2regulate the expression levels of cell wall invertases and sucrose synthases,which are involved in sucrose phloem unloading and sugar accumulation in fruits during grape berry development (Wanget al.2014).OsHXK7andOsHXK8are highly expressed in rice seeds during the starch-filling stage,andAtHXK3is highly expressed inArabidopsissink organs (roots and siliques) (Choet al.2006;Karveet al.2008). Furthermore,the expression ofMdHXK1,an orthologous gene ofAtHXK1-2in apple,increases in the early stages of apple fruit development and decreases after 60 days after blooming (DAB). Overexpression ofMdHXK1inArabidopsisresults in hypersensitivity to glucose,with the plants showing severely stunted cotyledons,hypocotyls,and roots.Moreover,MdHXK1promotes anthocyanin biosynthesis and improves salt tolerance by phosphorylatingMdbHLH3andMdNHX1,respectively,in apple (Huet al.2016;Sunet al.2018). Although some studies have been conducted on theMdHXK1gene function,the relationship betweenMdHXKgene family and the sugar content during fruit development had not been studied yet.
Apple (Malus×domesticaBorkh.) belongs to theMalusgenus in the Rosaceae family and has an autopolyploid origin. Whole-genome duplication (WGD) was essential in the chromosome number doubling from nine ancestral chromosomes to 17 chromosomes during apple domestication (Velascoet al.2010). After duplication,genes can become nonfunctional (gene death),neofunctional (novel function) or subfunctional (original function) (Rensing 2014). In general,most duplicated genes become nonfunctional due to unfavorable base mutations,while some duplicated genes maintain their original function,and some new genes evolve novel functions under positive selection (Blanc and Wolfe 2004;Panchyet al.2016). So far,few studies have reported on the evolutionary features of the hexokinase gene family,especially in apple. Hence,the evolutionary divergence and specific functions of duplicated genes during plant evolutionary processes need to be determined.
In this study,a total of nineMdHXKgenes were identified based on the recent apple resequencing genome GDDH13 (Daccordet al.2017). We evaluated their structures,subcellular localizations,chromosomal distributions,cis-element screening,phylogenetic relationships,gene duplication,evolutionary rates,positive selection and functional divergence. Additionally,the gene expression patterns and correlations between gene expression and sugar contents were also analyzed. Our results show the molecular evolution and functional characteristics of theMdHXKgene family in apple and provide a theoretical basis for future elucidation of the biological functions of theMdHXKgenes during apple fruit development.
2.Materials and methods
2.1.Identification of HXK genes and phylogenetic analyses
First,the protein sequences ofAtHXK1-3inArabidopsis(Karveet al.2008) were retrieved from The Arabidopsis Information Resource (TAIR) (https://www.arabidopsis.org/) and used as query sequences to identify the homologous genes of apple (Malus×domesticaBorkh.),pear (Pyruscommunis),peach (Prunuspersica) and black raspberry (Rubusoccidentalis) in the Genome Database for Rosaceae (GDR) (https://www.rosaceae.org/) by BLASTP (Chagnéet al.2014;VanBurenet al.2016;Daccordet al.2017;Verdeet al.2017). We then used BLAST 2.2.24 Software (http://blast.ncbi.nlm.nih.gov/Blast.cgi) to construct a BLAST database for each of these species,comparing amino acids versus amino acids. Finally,the putative orthologousHXKgenes were identified by reciprocal best similarity matching. These orthologousHXKgenes of the four Rosaceae species were used to perform phylogenetic analyses. ClustalX2 was used to perform multiple sequence alignments,and MEGA 7.0 was used to construct phylogenetic trees by the neighbor-joining (NJ) method (Kumaret al.2016).
2.2.Analysis of gene structure,conserved motifs,and subcellular localization
The gene structure of theMdHXKgenes was determined by Gene Structure Display Server 2.0 (http://gsds.cbi.pku.edu.cn/) based on their coding sequences and corresponding genomic sequences. The conserved motifs of the MdHXK proteins were analyzed by the MEME suite server (http://meme-suite.org/) based on their amino acid sequences,with the following software parameters:several output conserved motifs of 10,a maximum width of 50 amino acids and a minimum width of 10 amino acids. The conserved domains were then analyzed by the SMARTsuite server (http://smart.embl-heidelberg.de/). The subcellular localization of theMdHXKgenes was predicted by the PSORT II server (https://www.genscript.com/psort.html),which used theknearest neighbor (kNN) algorithm for assessing the probability of localization at each candidate site (Horton and Nakai 1997).
2.3.Screening of common and unique cis-elements in promoters
The upstream 2 000-bp promoter regions of the nineMdHXKgenes were retrieved from the Genome Database for Rosaceae (GDR;https://www.rosaceae.org/) and used to identify thecis-elementsviathe Plantcis-Acting Regulatory Element database (PlantCARE;http://bioinformatics.psb.ugent.be/webtools/plantcare/html/).Based on thecis-elements that were identified either in all theMdHXKgene promoters or in individualMdHXKgene promoters,we combined and eliminated the redundant replications among all thecis-elements and classified them into two categories:common and uniqueciselements.
2.4.Gene duplication and gene loss estimations
Based on the phylogenetic analysis of theMdHXKgenes,gene duplication and gene loss were estimatedviamanual examination in each subfamily. It was assumed that everyMdHXKgene subfamily should have at least one member from each Rosaceae species. We considered recent gene duplications to involve two or more genes from one species being classified adjacently in one subfamily,and old gene duplications to have occurred between two clades,in which each clade was identified as having at least one group ofHXKgenes from all four of the Rosaceae species. If theHXKgene of one Rosaceae species was not found in a subfamily,it was considered to represent a gene loss from that species.
2.5.Positive selection analysis of the four Rosaceae species
The ω values (theKa/Ksratio),which represented different evolutionary rates among pairs ofHXKgenes,were calculated in each subfamily by KaKs_Calculator Software 2.0 (https://sourceforge.net/projects/kakscalculator2/) with the maximum likelihood (ML) method (Wanget al.2010). Before calculation,the coding sequences of theHXKgenes were aligned by ClustalX2.Specifically,a pairwise comparison was used to measure the variation in ω values of theHXKgenes,which were selected randomly,among the different subfamilies of the phylogenetic tree. The ω values of each subfamily were used to analyze significant differencesviaSPSS 20.0 (SPSS Inc.,Chicago,IL,USA) and to estimate the evolutionary rates of each subfamily. In addition,by the use of the likelihood-ratio test (LRT) of the branch model,the ω values were obtained from Yang (2007). Briefly,the alignments of amino acid sequences and corresponding coding sequences were submitted to PAL2NAL (http://www.bork.embl.de/pal2nal/),and the output was used to estimate the ω values among different subfamilies of the phylogenetic tree using a branch-specific algorithm in the CodeML Program from PAML 4.9 (http://abacus.gene.ucl.ac.uk/software/paml.html) (Yang 2007). Alternativelikelihood models were then used to measure the evolutionary rates among the different subfamilies.
2.6.Plant materials and RNA sequencing
Five-year-old Golden Delicious apple (Malus×domesticaBorkh.) trees maintained at the Horticultural Experimental Station of Northwest A&F University,Yangling,Shaanxi Province,China,were used in this study. At 18 (cell division stage,as young fruits),34 (near the end of cell division),75 (early stage of cell expansion),95 (late stage of cell expansion),and 128 (end of sugar accumulation,as mature fruits) days after blooming (DAB) (Liet al.2016),fruit samples were taken at the same position of the trees between noon and 2:30 p.m.under full sun exposure. At each sampling time,five replicates of fruit samples,with at least eight fruits in each replicate from three trees,were harvested. The sampled fruits were immediately peeled,cut into small pieces,and frozen in liquid nitrogen. To compare the expression patterns of related genes in source and sink tissues,we also sampled shoot tips and mature leaves from the trees at 18 DAB. All frozen samples were stored at -80°C until their use for transcriptome sequencing and soluble sugar measurements. RNA-seq was performed using an Illumina HiSeq 2500 sequencer with 250 bp singleend sequencing (Illumina,San Diego,CA) at Biomarker Technologies,China. After removing barcode and adaptor sequences,the clean reads were aligned to theMalusgenome GDDH13,after which the raw counts of mapped reads for eachMalusgene model of GDDH13 were derived and normalized to the fragments per kilobase of transcript per million mapped reads (FPKM).
2.7.Measurements of soluble sugars and sorbitol contents in apple fruits
As we previously described (Liet al.2018),soluble sugars and sorbitol were extracted and derivatized sequentially with methoxyamine hydrochloride and N-methyl-Ntrimethylsilyl-trifluoroacetamide (MSTFA). Briefly,100 mg of mixed samples was extracted by 1.4 mL of 75% methanol,with 100 μL of ribitol used as an internal standard. After fractionation of nonpolar metabolites by chloroform,2 and 20 μL aliquots of the polar phase were transferred to 1.5-mL centrifuge tubes for assessing the highly abundant metabolites (such as sorbitol and sucrose) and less abundant metabolites (such as glucose,fructose,and galactose),respectively. They were then dried under vacuum and derivatized with methoxyamine hydrochloride and MSTFA sequentially. Afterward,the metabolites were analyzed with a Shimadzu GCMS-2010SE (Shimadzu Corporation,Kyoto,Japan) and quantified based on standard curves generated for each metabolite and internal standard.
3.Results
3.1.Identification and characterization of the HXK gene family in apple
In this study,we identified a total of nine putativeMdHXKgenes in the GDDH13 apple genome (Daccordet al.2017);their coding sequences and amino acid sequences are shown in Appendix A. Furthermore,the physiological and biochemical properties of the nine MdHXK proteins were analyzed by ProtParam (https://web.expasy.org/protparam/),and the summary information is shown in Table 1. The nineMdHXKgenes were located on six different chromosomes.The lengths of the MdHXK proteins ranged from 492 (MD15G1012900) to 532 (MD02G1194700) amino acids,with predicted molecular weights (MWs) of 53.12 to 58.23 kDa,respectively. The theoretical isoelectric points (pI) of the MdHXK proteins ranged from 5.38 (MD15G1012900) to 7.41 (MD11G1188100),and the grand averages of hydropathicity (GRAVY) ranged from -0.295 (MD02G1194700) to 0.011 (MD11G1188100).In addition,the instability index values of the MdHXK proteins ranged from 28.32 (MD17G1183100) to 44.66 (MD15G1012900),and the aliphatic index values ranged from 83.55 (MD02G1194700) to 99.86 (MD17G1183100).

Table 1 Summary information of physiological and biochemical properties of MdHXK proteins1)
3.2.Chromosomal localization and duplication of MdHXK genes
To investigate the genomic distribution of theMdHXKgenes,their positions on the apple genome chromosomes were mapped. The chromosomal localization of the nineMdHXKgenes is shown in Fig.1. Among these genes,four were distributed on two homologous pairs of chromosomes (MD03G1170700/MD11G1188100 on Chr3/Chr11,MD09G1202200/MD17G1183100 on Chr9/Chr17),two (MD02G1066500 and MD02G1194700) were located on chromosome 2,and three (MD15G1012900,MD15G1197000 and MD15G1305100) were located on chromosome 15.
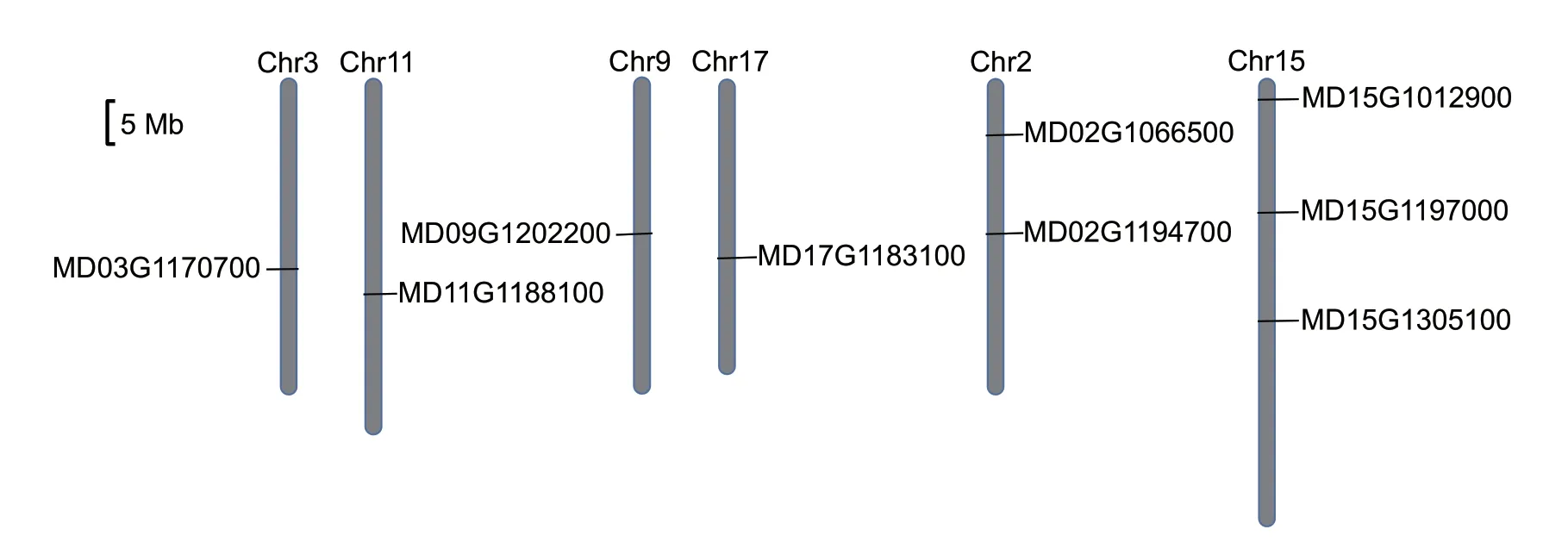
Fig.1 Chromosomal localization of the MdHXK genes in apple.
Gene duplication,including whole-genome duplication (WGD),segmental duplication,and random duplication,has long been considered an important evolutionary driver for animals,fungi,and other organisms,especially plants,and is considered to have played a vital role in the process of apple domestication (Maet al.2018a,b). To determine the evolutionary rates and selective pressures among theMdHXKgenes and their duplicated genes,we usedKa(nonsynonymous),Ks(synonymous),and theKa/Ksratio (ω) for estimations. In general,ω<1 represents purifying selection;ω=1 represents neutral evolution;and ω>1 represents positive selection (Blanc and Wolfe 2004;Panchyet al.2016). We calculated the selective pressures of each duplicated gene pair,and the ω values are shown in Appendix B. Interestingly,the ω values of all theMdHXKgene pairs were less than 1,which indicated that theMdHXKgene family underwent strong purifying selection and demonstrated that this family was conserved during apple domestication.
3.3.Gene structure,conserved motifs,and subcellular localization of MdHXK genes
To better understand the gene structure of theMdHXKgenes,exon-intron diagrams were analyzed by the Gene Structure Display Server (http://gsds.cbi.pku.edu.cn/) according to the coding sequences and genomic sequences (Fig.2-A). The results showed that each of theMdHXKgenes contained nine exons separated by eight introns. Similar exon-intron structures ofHXKgenes have been reported in other plant species,including pear,rice,andArabidopsis,suggesting that the genomic structures of members of theHXKgene family are evolutionarily conserved across plant species (Choet al.2006;Karveet al.2008;Yuet al.2018). Moreover,two pairs of paralogous genes from apple,MD03G1170700/MD11G1188100 and MD09G1202200/MD17G1183100,presented similar exon and intron lengths,which suggested that these genes might originate from duplication of their paralogous counterparts.
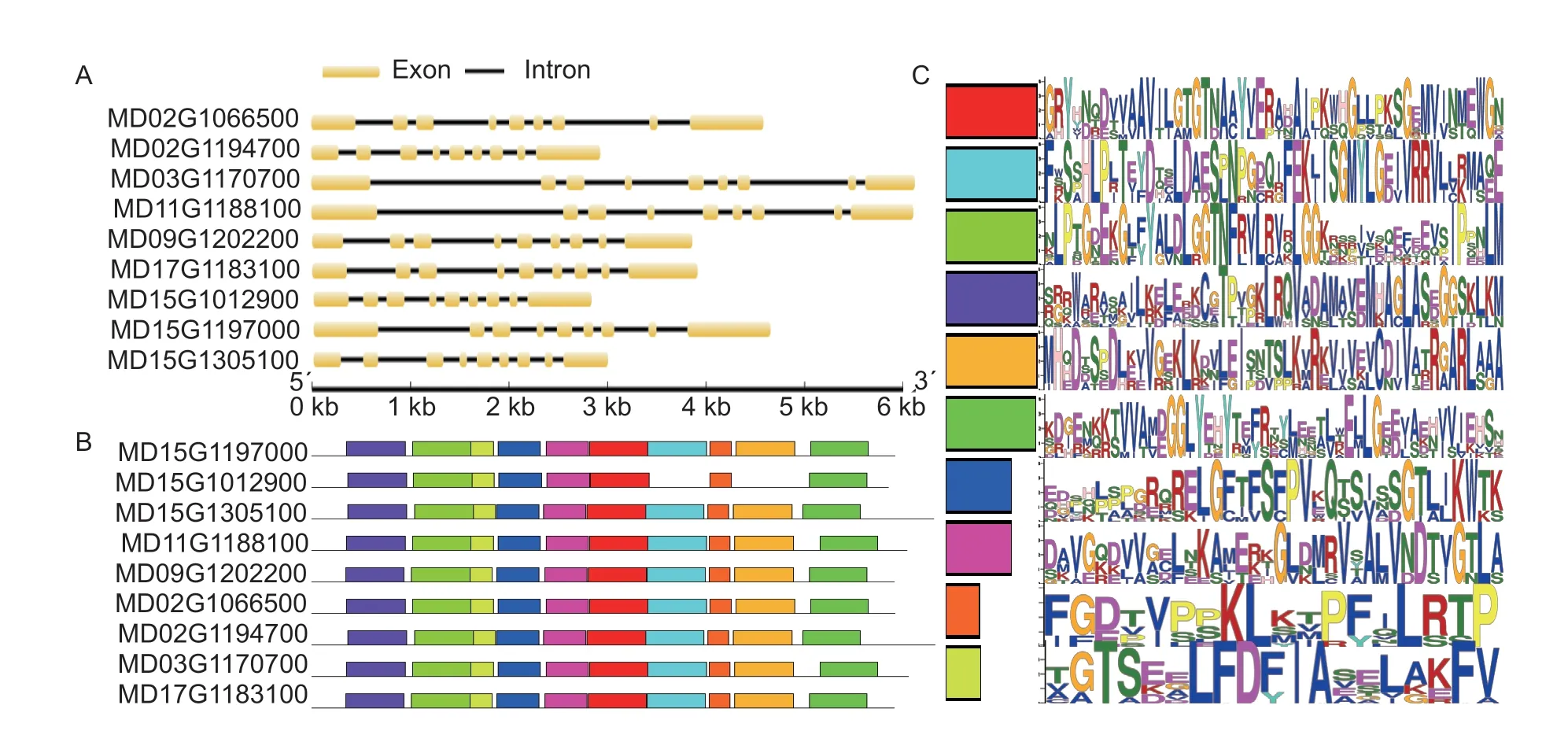
Fig.2 The gene structure and conserved motifs of MdHXK genes in apple. A,the gene structure of MdHXK genes,in which yellow lines represent exons and black lines represent introns. B,the conserved motifs of MdHXK proteins,in which the conserved motifs are indicated by colored boxes. C,sequences of the 10 conserved motifs in the MdHXK proteins.
To further systematically identify the motifs of the MdHXK proteins,the MEME server (http://meme-suite.org/) was used to estimate the distribution of conserved motifs. The results showed that 10 putative conserved motifs were identified in all MdHXK proteins except MD15G1012900,for which the gene lacked two motifs (Fig.2-B),and the lengths of the conserved motifs ranged from 17 to 50 amino acids (Fig.2-C). In addition,all of the MdHXK proteins contained two conserved domains of Hexokinase_1 and Hexokinase_2,and some of them contained transmembrane regions and signal peptides (Appendix C). Thus,theMdHXKgenes clearly exhibited extreme conservation during the evolutionary process.
Subcellular localization of the MdHXK proteins was predicted using the website PSORT (https://www.genscript.com/psort.html). The prediction results showed that apple MdHXK proteins are found in most subcellular structures,including the mitochondria,cytoplasm,nucleus,and endoplasmic reticulum. Of these nine MdHXK proteins,six displayed similar localization patterns and were mainly located in the nucleus,cytoplasm and mitochondria,with relative values of 34.8,21.7,and 21.7%,respectively. Similarly,MD02G1066500 was also mainly located in the nucleus,cytoplasm,and mitochondria,but the greatest abundance was in the mitochondria (30.4%). In addition,MD15G1012900 had the largest predicted value in mitochondria,reaching 91.3% (Table 2). These results indicated that the MdHXK proteins not only sense sugar starvation as signaling molecules but also participate in the glycolytic pathway to provide energy for plant growth and development.
3.4.Analysis of the cis-elements in MdHXK promoters
To predict the functions of theMdHXKgenes,2-kb promoter regions upstream of the initiation codons of theMdHXKgenes were analyzed using PlantCARE website (http://bioinformatics.psb.ugent.be/webtools/plantcare/).A total of 52cis-elements were identified in the promoter regions of all studiedMdHXKgenes (Appendix D),and most of them responded to light,hormones,abiotic stress,and so on. Differentcis-elements indicate different functions. In this study,we identified some commoncisacting elements that existed in most of theMdHXKgene promoters. Among thesecis-elements,five are responsive to light,and four are responsive to hormones,such as methyl jasmonate (MeJA),abscisic acid (ABA),gibberellin,and salicylic acid (SA). The presence of the drought stress-responsive element MYC and the abiotic stressresponsive element STRE indicate thatMdHXKgenes may respond to abiotic stress. The presence of a sugar metabolism signaling element W-box indicates thatMdHXKmay function as a sugar cleavage catalyst and may sense sugar signals (Table 3). In addition,we identified eight uniquecis-acting elements distributed within sixMdHXKgene promoter regions. Twocis-elements,Gap-boxes and LAMP-elements,are responsive to light,the AT1-motif element is responsive to auxin,and the threeciselements are gene-specific binding sites (Table 4). Hence,we considered that the uniquecis-acting element may be responsible for the different functions.
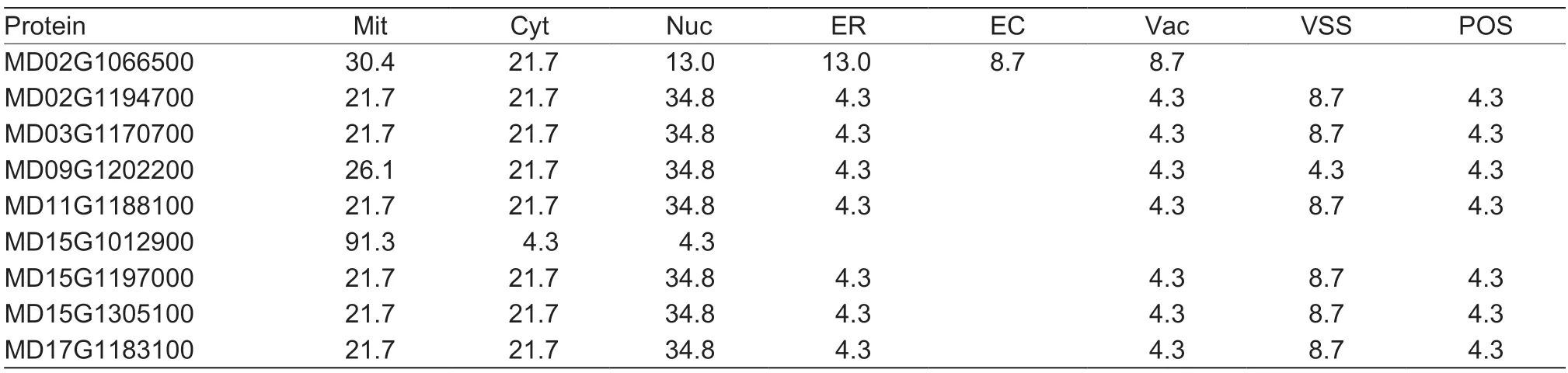
Table 2 Prediction of subcellular localization of the nine MdHXK proteins (%)1)
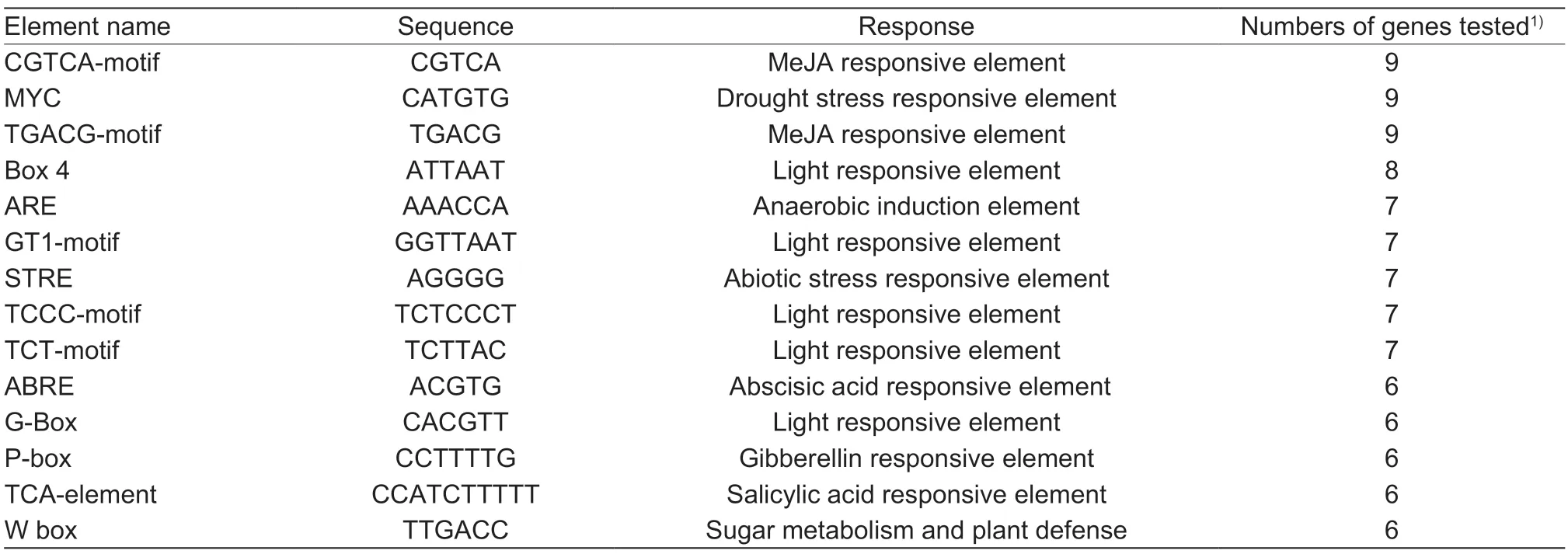
Table 3 Common cis-acting elements identified in the MdHXK genes
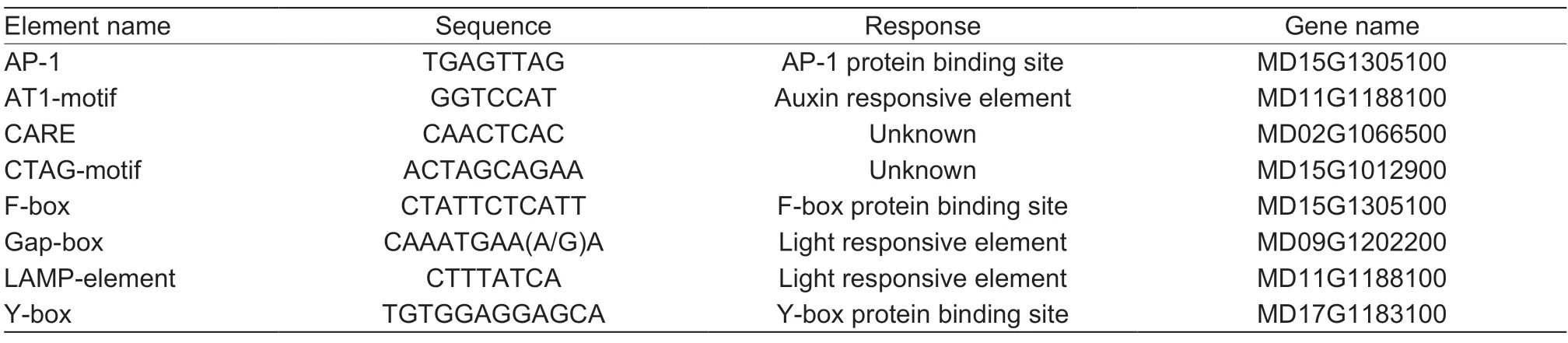
Table 4 Unique cis-acting elements identified in the MdHXK genes
3.5.Phylogenetic analysis,gene duplication,and gene loss of the MdHXK genes in four Rosaceae species
To determine the evolutionary relationships of hexokinase genes among Rosaceae plants,we chose four species,apple (Malus×domesticaBorkh.),pear (Pyruscommunis),peach (Prunuspersica) and black raspberry (Rubusoccidentalis),to construct a phylogenetic tree of the HXKs using the neighbor-joining (NJ) method. In this analysis,nine,nine,seven and fiveHXKgenes were identified in the apple,pear,peach and black raspberry genomes,respectively,and the sequence information for all the HXK proteins were downloaded from the Genome Database for Rosaceae (https://www.rosaceae.org/). According to the evolutionary relationship shown in Fig.3,all HXK proteins clustered into five subfamilies,and each Rosaceae species gene was clearly divided into each subfamily;the bootstrap value of each subfamily was more than 50%.Moreover,of the nineMdHXKgenes,two,two,two,one and two genes were classified into the I-V subfamilies,respectively (Fig.3).
Gene duplications can be divided into two categories,old duplications and recent duplications,based on the evolutionary history of Rosaceae plants (Taylor and Raes 2004). We considered that old duplications represented gene duplications that occurred before the radiation of Rosaceae plants,whereas recent duplications represented gene duplications that occurred in the same Rosaceae species (Liet al.2010;Wanget al.2016).In this study,we identified three old duplication events (Fig.3,red triangles) and two recent duplication events (red squares),indicating that all theHXKgenes except the two sister pairs ofHXKgenes,Prupe.1G366000.1-Prupe.1G366000.2 and Prupe.4G256200.1-Prupe.4G256200.2,existed prior to the radiation of the Rosaceae species. Interestingly,we found that an old duplication event existed within another old duplication event,suggesting that a gene duplication event had occurred before the Rosaceae species radiation. In addition,gene loss was not detected in these four Rosaceae species (Fig.3).
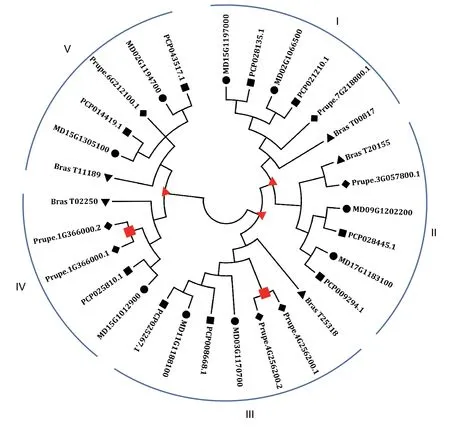
Fig.3 Phylogenetic tree analysis of the HXK genes in four Rosaceae species. Recent and old duplications are indicated by red squares and triangles,respectively. Bootstrap values of all branches are higher than 50%.
3.6.Positive selection analysis of the HXK genes in four Rosaceae species
To determine the evolutionary rates ofHXKgenes in each subfamily,the pairwise ω values for all identifiedHXKgenes in each subfamily were calculated using a pairwise comparison model (Fig.4;Appendix E) (Blanc and Wolfe 2004;Panchyet al.2016). We found that all ω values among each subfamily were less than 1,indicating that allHXKgenes underwent purifying selection and that there was no positive selection. In addition,comparing the mean ω values of theHXKgenes in each subfamily gave the following values:subfamily V>subfamily III>subfamily I>subfamily II>subfamily IV (Fig.5). TheHXKgenes from subfamily III and subfamily V had significantly higher ω values than those from the other subfamilies,demonstrating that theHXKgenes from these two subfamilies exhibited a significant difference in evolutionary rates.
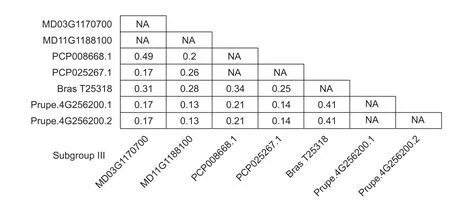
Fig.4 Estimation of ω values of HXK gene subfamily III using the ML method.
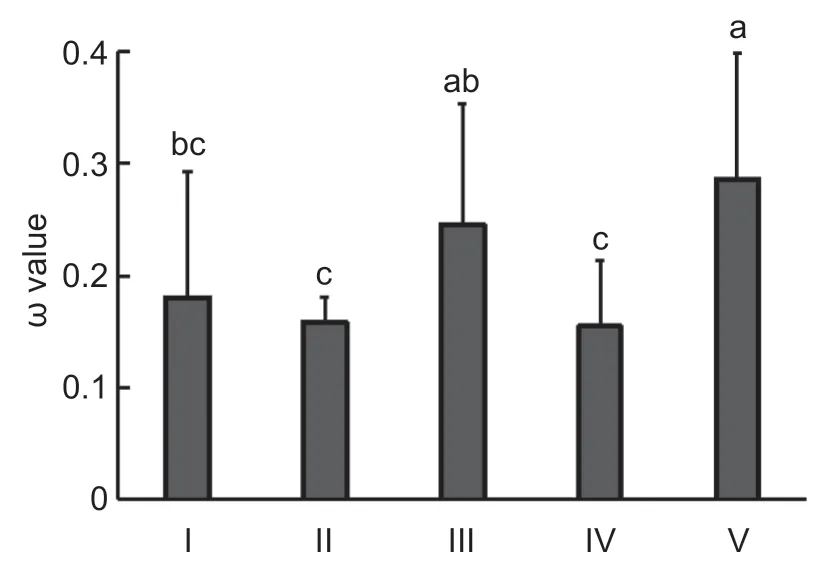
Fig.5 Mean ω values of the HXK genes in different subfamilies.Different lowercase letters indicate significant differences among subfamilies (t-test,LSD test at P<0.01). Error bars correspond to SE of means.
Moreover,to better understand the gene evolutionary rates of the five HXK subfamilies,that is,which subfamily had diverged from the most recent common ancestor,the ω values between old duplicated subfamilies were calculated again by the branch-specific model in PAML.As shown in Table 5,the two-ratio model was favored in two pairs of HXK groups,I and II,as well as I+II and III,both of which hadP-values less than 0.05,indicating that the ω values of subfamilies II and III differed significantly from those of subfamilies I and I+II,respectively.However,theP-values of the other two groups were greater than 0.05,suggesting that they had similar evolutionary rates during apple domestication.
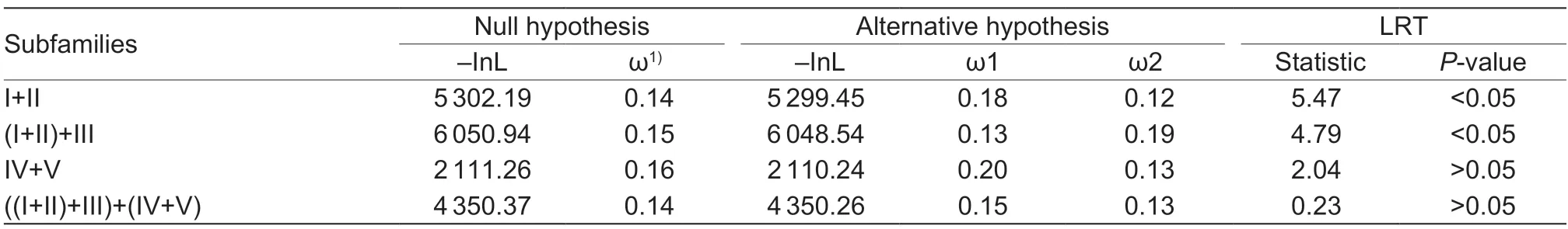
Table 5 Likelihood-ratio test (LRT) statistics and parameters from Branch model of PAML
3.7.Expression analysis of the MdHXK genes in source and sink tissues of apple plants
Hexokinase is capable of phosphorylating hexose molecules (glucose and fructose) and is the first irreversible catalytic enzyme in the glycolytic pathway.Hexokinase can provide energy for plant growth and development,and its expression is more active in rapidly growing tissues (Liet al.2012). To better understand the source-sink expression patterns ofMdHXKgenes in apple plants,RNA-seq was used to evaluate the expression levels of theMdHXKgenes in four tissues:shoot tips (STs),mature leaves (MLs),young fruits (YFs),and mature fruits (MFs). As sink tissues,shoot tips,and young fruits accept sugars from the outer source tissues,which are rapidly metabolized for rapid growth and development,andMdHXKgenes may be involved in the phosphorylation of hexoses and the provision of energy in these two tissues. Mature leaves are a carbohydrate-producing source tissue,andMdHXKgenes may affect photosynthesis and sugar loading by sensing sugar signals. Cells of mature fruits are replete with hexoses and sucrose,which may be involved in the functions of theMdHXKgenes.
The nineMdHXKgenes showed significantly different expression patterns among the four kinds of apple tissues. In the transcriptome data,the FPKM value of MD15G1197000 was higher than those of other genes in all four tissues (Appendix F). In contrast,MD02G1194700 and MD15G1305100,which belong to subfamily V,were expressed at low levels in all four tissues,with relative expression levels ranging from 0-0.98 and from 0-0.02,respectively (Fig.6;Appendix F). Additionally,a pair of paralogous genes,MD09G1202200 and MD17G1183100,had similar expression patterns,with the highest expression in fruits,the second highest in shoot tips,and the lowest in mature leaves. Similarly,fourMdHXKgenes exhibited the highest expression levels in the mature leaves and mature fruits. Furthermore,six of the nineMdHXKgenes had the highest expression levels in mature fruits,indicating that these genes may play important roles in apple fruits.

Fig.6 The expression patterns of the MdHXK genes in four source and sink tissues. ST,shoot tips;ML,mature leaves;YF,young fruits;MF,mature fruits. The heat map was constructed based on relative expression levels. Different colors represent different expression levels,with red representing the highest level.
3.8.Analysis of the expression of MdHXK genes at different developmental stages of apple fruits
To explore the expression patterns of the members of theMdHXKgene family in different stages of apple development,RNA-seq was used to measure the expression levels of the nineMdHXKgenes at five different fruit stages:18,34,75,95,and 128 DAB. The expression patterns of theMdHXKgene family members exhibited significant differences among the five stages.MD15G1305100 was not detected in any of the five stages,while MD02G1194700 was detected only at two stages:95 and 128 DAB. Additionally,the expression levels of three genes,MD03G1170700,MD11G1188100,and MD02G1066500,increased with the development of apple fruits. In contrast,the expression levels of three other genes,MD15G1012900,MD15G1197000,and MD09G1202200,had negative correlations with fruit development. Furthermore,the expression level of MD17G1183100 decreased after 34 DAB but increased after 95 DAB (Fig.7;Appendix G).

Fig.7 The expression patterns of the MdHXK genes in five developmental stages of apple fruit. DAB,days after blooming.The heat map was constructed based on relative expression levels. Different colors represent different expression levels,with red representing the highest level.
With the development of apple fruits,the sugar contents change dynamically. The glucose concentration decreased from 18 to 75 DAB and then increased toward maturity,whereas the fructose concentration increased from 18 to 95 DAB and decreased slightly at maturity. The sucrose concentration increased nearly 7.5-fold from 18 DAB to maturity,but the sorbitol concentration decreased with fruit development. The galactose concentration pattern was similar to that of glucose. Overall,the total sugar content increased from 18 to 95 DAB and remained unchanged until maturity (Fig.8;Appendix H).
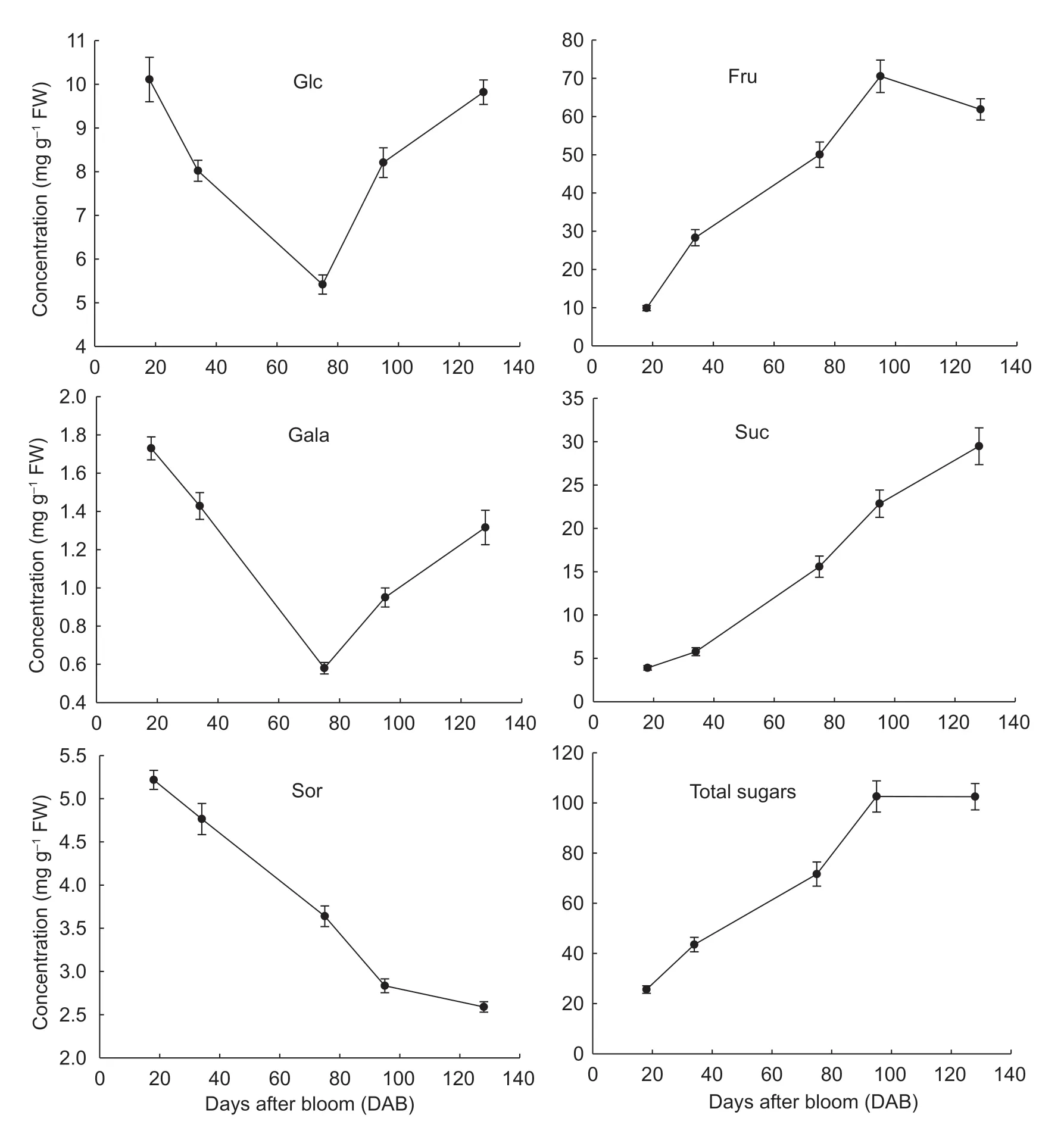
Fig.8 Concentrations of glucose (Glc),fructose (Fru),galactose (Gala),sucrose (Suc),sorbitol (Sor) and total sugars during apple fruit development. Values are means of five replicates±SD.
To determine whether the expression of theMdHXKgenes is related to sugar accumulation in apple fruits,the Pearson correlation coefficients (PCCs) were analyzed.As shown in Table 6,the expression of MD11G1188100 had a significant positive correlation with fructose concentration and a negative correlation with galactose concentration. In addition,two genes,MD02G1066500 and MD02G1194700,had significant positive correlations with sucrose concentration,and the correlation with the former was extremely significant. The expression of MD09G1202200 and MD15G1012900 was significantly negatively correlated with sucrose content,but positively correlated with sorbitol content. Furthermore,MD02G1066500 had a significant negative correlation with sorbitol content,but a positive correlation with the total sugar concentration. Lastly,MD09G1202200 was significantly negatively correlated with the total sugar content (Table 6).
4.Discussion
HXKgenes play important roles not only in providing energy for plant growth and development through the phosphorylation of hexoses but also as signaling agents that sense carbon starvation in the cells (Junget al.1997;Granot 2007). To date,members of theHXKgene family have been identified in many species,including six members inArabidopsis,ten in rice,nine in maize,seven in cassava,and ten in pear (Choet al.2006;Karveet al.2008;Zhanget al.2014;Genget al.2017;Yuet al.2018). The recent high-qualityde novoassembled apple reference genome GDDH13 (Daccordet al.2017),which was constructedviathird-generation transcriptome sequencing technology,served as a convenient framework for analyzing theMdHXKgene family. We identified a total of nineMdHXKgenes in the apple genome (Table 1),and our results revealed more members than had been previously reported (Liet al.2012). We think the reason for this difference is that the new genome has more identified and mapped genes than the previous version.
Apple (Malus×domesticaBorkh.) is a diploid species that has an autopolyploid origin and 17 chromosomes (Velascoet al.2010). Previous research has shown that the apple genome underwent whole-genome duplication (WGD),segmental duplication,and random duplication during the domestication of apple,and these are the most important types of gene duplication events (Velascoet al.2010;Maet al.2018a,b). In our study,among the nineMdHXKgenes,four were located on two homologous pairs of chromosomes (Fig.1). For example,the two homologous chromosomes 9 and 17 each contained anMdHXKgene in the middle. Similar findings were also obtained for homologous chromosomes 3 and 11.These results demonstrated that the apple genome had undergone WGD. Additionally,there were two and threeMdHXKgenes on chromosomes 2 and 15,respectively,indicating that segmental duplication might have occurred on these two chromosomes. However,noMdHXKgenes were detected in the homologous chromosomes of chromosome 2 or 15,which might indicate gene loss and divergence after WGD.
To determine the evolution of theMdHXKgenes,their gene structures and conserved motifs were characterized. All theMdHXKgenes exhibited the same exon-intron index,with structures similar to those in other species (Choet al.2006;Karveet al.2008;Zhanget al.2014;Genget al.2017;Yuet al.2018). Moreover,ten putative conserved motifs were identified in all MdHXK proteins except MD15G1012900,which lacked two motifs (Fig.2). These results clearly indicated that theMdHXKgenes were extremely conserved during the evolutionary process.AtHXK1is a dual-function enzyme inArabidopsis,which is localized in both the cytoplasm and nucleus. It exhibits catalytic activity in the glycolytic pathway and also functions in the sugar sensing and signaling pathways (Choet al.2006;Chenget al.2011).AtHXK1mutations with little or no catalytic activity have been shown to mediate diverse Glc responses (Harrington and Bush 2003). Furthermore,theHXKslocalize to the nucleus and can modulate specific target gene transcription independent of Glc metabolism (Choet al.2006). The predicted subcellular localization showed that nearly all of the MdHXK proteins were most abundant in the mitochondria,cytoplasm,and nucleus (Table 2),confirming thatMdHXKhas dual functions:catalyzing the breakdown of hexose molecules and acting in signal transduction. In general,different ω values represent different evolutionary rates (Wanget al.2016). The ω values of all theMdHXKgene pairs were less than 1 (Appendix B),indicating that theMdHXKgene family had a slow evolutionary rate and underwent strong purifying selection.
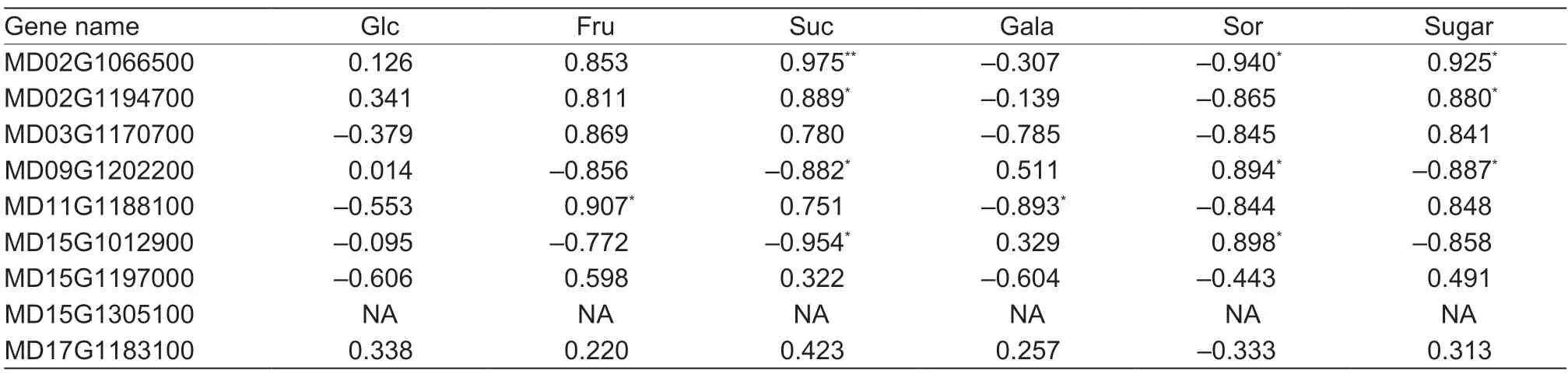
Table 6 Pearson correlation coefficient (PCC) between MdHXK expression and sugar contents of apple fruit during development1)
In addition,phylogenetic analysis of theMdHXKgenes among four Rosaceae species resulted in their clustering into five subfamilies (Fig.3;Appendix I). We detected two recent duplications in peach and three old duplications in Rosaceae species. There were 9,9,7 and 5HXKgenes (before recent duplication),accompanied by 17,17,8,and 7 chromosomes in apple,pear,peach,and black raspberry,respectively. This nearly doubled relationship ofHXKgene numbers indicated that apple and pear experienced a WGD event but that peach and black raspberry did not,which is consistent with the results of a previous study (Velascoet al.2010;Wuet al.2013).Interestingly,everyMdHXKgene was grouped with a pearHXKgene,indicating that apple and pear had the closest genetic relationship among the Rosaceae species in the analysis. Furthermore,the ω values of allHXKgene pairs within each subfamily were less than 1 (Appendix E),indicating that all theHXKgenes underwent purifying selection and that no positive selection had occurred.However,the average ω values of subfamilies III and V were higher than those of the other families,and thePvalues of subfamilies II and III were significantly different from those of subfamilies I and I+II (Table 5),indicating different evolutionary rates among each subfamily.
Tissue-specific expression patterns can provide a better understanding of gene function in plants (Genget al.2017;Maet al.2019). In this research,theMdHXKgenes also showed diverse expression patterns in four different source-sink tissues. Of the nineMdHXKgenes,seven genes were expressed in all the tissues evaluated,but two genes,MD02G1194700 and MD15G1305100,were expressed in only some of the tissues. In addition,four genes,MD02G1066500,MD11G1188100,MD03G1170700,and MD15G1197000,had the highest expression levels in the mature leaves and mature fruits,while two genes,MD09G1202200 and MD17G1183100,were expressed at the highest levels in the fruits (young fruits and mature fruits). However,the expression pattern of only one gene,MD15G1012900,was different from that of theMdHXKgenes. The changes in gene promoter regions may affect gene expression levels. For example,the promoter ofMdMYB10contains five direct tandem repeats of a 23-bp sequence,which increases the transcript level ofMdMYB10and promotes the coloring of apple peel (Espleyet al.2009). Therefore,we considered that the difference in MD15G1012900 expression found here might be due to the lack of two conserved motifs and presence of uniquecis-elements in the promoter (Figs.2 and 6;Table 4). TwoMdHXKhomologous genes,MD03G1170700 and MD11G1188100,which were located on the homologous pairs of chromosomes 3 and 11,had the highest expression levels in the mature leaves and mature fruits,respectively,although they had similar expression patterns. Similarly,another pair ofMdHXKhomologous genes,MD09G1202200 and MD17G1183100,had the highest expression levels in the young fruits and mature fruits,respectively (Fig.6;Appendix J). These results indicated that gene duplication ofMdHXKscould have led to the expression differences and functional divergence.
Sugar content changes dynamically with apple fruit development (Liet al.2012). Interestingly,we found that the expression of three genes,MD03G1170700,MD11G1188100 and MD02G1066500,had a significant positive correlation with the fruit developmental stage,suggesting that these genes may be more closely related to the modulation of glycolytic flux and energy supply of the cells of young fruits. Four genes,MD15G1012900,MD15G1197000,MD09G1202200,and MD17G1183100,had significant negative correlations with fruit developmental stage (Fig.7),suggesting that these genes may be related to the fast utilization of glucose released from starch breakdown (Claeyssen and Rivoal 2007;Karve and Moore 2009;Liet al.2012). The Pearson correlation coefficients (PCCs) showed significant correlations between some of theMdHXKgene expression levels and hexose,sucrose,or total sugar contents,indicating that theMdHXKgenes might affect sugar accumulation in apple fruits (Table 6). None of theMdHXKgenes showed a significant correlation with glucose concentration,and we considered that multipleMdHXKgenes or genes from other pathways co-regulated the glucose contents in apple fruits.
5.Conclusion
In this work,we provide a deep systematic understanding of the evolutionary pattern ofMdHXKgenes in apple (Malus×domesticaBorkh.). A total of nineMdHXKgenes were identified in the recently published apple genome,GDDH13. All theMdHXKgenes had similar gene structures,conserved motifs,and subcellular localizations.There are various common and uniquecis-elements in theMdHXKpromoter regions. The chromosomal locations of theMdHXKgenes showed that genome-wide duplication,segmental duplication,and tandem duplication occurred during apple domestication. Furthermore,theKa/Ksratio of theMdHXKgene pairs indicated that theMdHXKgenes had undergone strong purifying selection. Moreover,a phylogenetic tree ofHXKgenes was constructed from four Rosaceae species,and five subfamilies were classified. Based on the phylogenetic relationships,two recent duplication events,three old duplication events,and significant differences in evolutionary rates among the HXK subfamilies were identified. Additionally,divergent expression patterns of the nineMdHXKgenes in four source-sink tissues at five different developmental stages of apple fruits were detected. In general,our results indicate thatMdHXKgenes play vital roles in apple fruit development and sugar accumulation.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31672128) and the Training Program Foundation for the Young Talents of Northwest A&F University,China (2452020004).
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendicesassociated with this paper are available on http://www.ChinaAgriSci.com/V2/En/appendix.htm
杂志排行

Journal of Integrative Agriculture的其它文章
- Lignin metabolism regulates lodging resistance of maize hybrids under varying planting density
- Adoption of small-scale irrigation technologies and its impact on land productivity:Evidence from Rwanda
- Comparison of grain yield and quality of different types of japonica rice cultivars in the northern Jiangsu plain,China
- Natural nematicidal active compounds:Recent research progress and outlook
- lmproving grain appearance of erect-panicle japonica rice cultivars by introgression of the null gs9 allele
- Comparative transcriptome analysis of different nitrogen responses in low-nitrogen sensitive and tolerant maize genotypes