中国襀翅目昆虫DNA条形码技术研究
2021-06-16陈志腾
陈志腾
(江苏科技大学 粮食学院,镇江 212100)
襀翅目(Plecoptera)昆虫,俗名石蝇(stonefly),是一类重要的半变态类水生昆虫,其稚虫常栖息于清澈的流水中,老熟稚虫漂浮至水面或爬上岸边羽化成虫. 该目昆虫由于对水质变化极为敏感,因此是水质监测的重要生物指标. 近年来,由于我国城市化进程显著加快,各流域的水质受到了不同程度的影响. 大量的襀翅目昆虫正在快速灭绝,其中甚至包括尚未被发现和描述的新物种. 由于传统的形态学鉴定方法耗时、耗力,且需要专业的分类学技能,所以迫切需要寻找一种快速鉴定的方法,来协助水质监测工作和生物多样性的调查.
DNA条形码技术是近十几年来逐渐发展起来的一项技术,该技术通过线粒体基因组中COI基因的部分片段来实现物种的快速鉴定[1]. 目前,国外已经有研究借助DNA条形码技术对襀翅目昆虫进行快速鉴定、不同生长期的匹配,以及种群遗传学方面的研究[2-4]. 同时,也有研究建立了当地的襀翅目DNA条形码数据库[5-7]. 目前已知的中国襀翅目DNA条形码序列大多来源于线粒体基因组数据,仅有少数来源于将形态学与基因结合的分类学研究[8-9].但是,中国襀翅目的DNA条形码尚无专门研究,这个空缺阻碍了该类群的深入研究.
为了对中国襀翅目的遗传特征有更进一步的了解,文中统计了中国襀翅目DNA条形码数据,利用生物信息学方法分析了DNA数据的遗传特征,并基于DNA条形码数据进行了系统发育分析,验证了这些数据在物种划分和亲缘关系重建方面的作用,为中国襀翅目快速鉴定和遗传多样性研究提供了参考.
1 实验方法
1.1 中国襀翅目DNA条形码数据获取
以GenBank (https://www.ncbi.nlm.nih.gov/)数据库和BOLD条形码数据库(http://www.boldsystems.org/)进行在线襀翅目COI基因搜索,发现中国襀翅目36个物种已经测序,但是仅有28个物种的42条COI基因序列已公开(表1). 将该42条基因序列以.fasta文件格式下载并存储.
1.2 DNA条形码分析及系统进化树构建
利用ClustalX 1.83程序对获取的DNA序列进行多序列比对分析,序列比对结果通过Mega 6.0程序进行核苷酸成分分析、位点分析和基于K2P模型的遗传距离分析. 使用DAMBE 4.2.13程序对序列中核苷酸替换(转换/颠换)的饱和性进行分析. 使用Mega 6.0程序的邻接法 (Neighbor-joining NJ) 构建中国襀翅目DNA条形码的系统进化树,设置参数如下:自举值为1000,模型为kimura-2-parameter,碱基替换模型为transitions+transversions,位点进化速率为uniform rates,空缺数据删除方法为pairwise deletion. Mega 6.0程序导出的Newick格式树文件通过FigTree 1.4.3 程序进行可视化分析,自展分析的各节点支持率标记在进化树节点上.
2 结果与讨论
2.1 中国襀翅目DNA条形码数据统计
文中获取的DNA条形码序列来源于28种、26属、8科(表1),主要集中于襀科、叉襀科和卷襀科. 而大襀科、带襀科的DNA条形码仍然空缺. 因此,这些条形码的数量与各科的物种数量呈正相关关系,物种数较多的科,往往具有较多DNA条形码数据,而那些物种稀少的科,则具有较少的数据(图1).

图1 中国襀翅目各科DNA条形码已知物种数
2.2 中国襀翅目DNA条形码特征
根据ClustalX 1.83程序比对分析,DNA条形码序列的重叠长度为619 bp,足够用于物种的快速鉴定(图2). 经过计算,所有序列都明显偏向于使用A碱基和T碱基,表现出偏高的A+T含量:平均A+T含量为60.0%,最高A+T含量为64.8%,最低为53.5% (表2).

表2 中国襀翅目DNA条形码的碱基成分
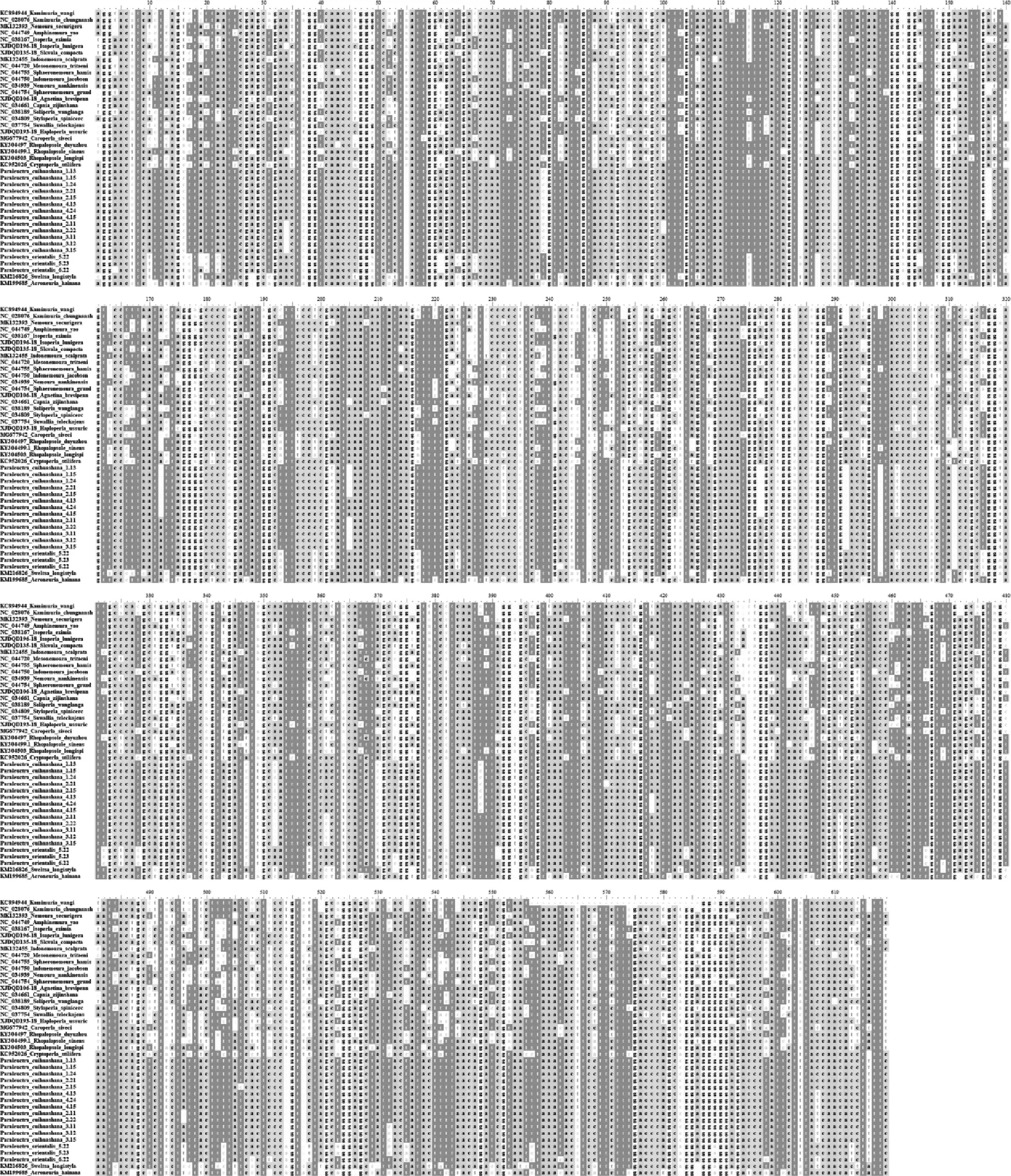
图2 中国襀翅目DNA条形码序列比对
在这些条形码序列中,共检测到360个未发生碱基变异的保守位点(Conserved sites),占重叠序列的58.2%. 此外,还检测到259个存在2种或2种以上碱基变异的变异位点(Variable sites),占重叠序列的41.8%. 这些条形码的核苷酸序列可以转换成206个氨基酸序列,其中丝氨酸Ser出现频率最高,而谷氨酸Glu出现频率最低.
DAMBE程序的核苷酸替代饱和性分析结果中(表3),ISS 表3 DNA序列替换饱和度检验 基于Kimura-2-parameter (K2P)模型和COI基因序列,28种襀翅目昆虫的遗传距离经计算如图3. 在28种襀翅目昆虫内,遗传距离变化较大,最大为30%. 对于同一物种来说,种内不同个体之间的遗传距离较低,最小为0,最大为4%. 对于同属内的不同物种来说,遗传距离变化较大,最小为0,最大为24%. 这两个分类水平的遗传距离范围有所重叠,说明常用的遗传距离阈值2%不完全适用于襀翅目所有类群. 应当进行更多的采样和测序,对不同分类阶元的遗传距离阈值进行分析,从而选择出最优的一系列阈值用于以后的DNA条形码研究. 在基于DNA条形码序列构建的邻接树中(图4),翠华山拟卷襀(Paraleuctracuihuashana)和东方拟卷襀(Paraleuctraorientalis)两个具有多于1条序列的物种的单系性得到了强烈支持,且节点支持率较高. 拟卷襀属(Paraleuctra)、诺襀属(Rhopalopsole)和钩襀属(Kamimuria)均分别聚为单系分支,与形态学鉴定结果一致. 但是球尾叉襀属(Sphaeronemoura)、黑襀属(Capnia)、印叉襀属(Indonemoura)、叉襀属(Nemoura)和同襀属(Isoperla)的单系性未得到支持. 这些单系性未被支持的类群由于可用的DNA条形码序列有限,无法与相近的类群区分开. 同时,在线数据库中也可能存在错误鉴定的种类,测序产生的错误序列对系统进化树的拓扑结构造成了不良影响. 因此,需要在以后的研究中通过大量测序相同物种的不同个体,经过序列比对分析保留序列相似的物种,而排除那些具有明显差异的序列. 在更高的科级水平,DNA条形码序列由于碱基数量有限,碱基所代表的遗传信息也很局限,无法将各科有效区分. 图4 基于DNA条形码构建的中国襀翅目邻接树 研究表明,基因水平的鉴定与分析对生物的系统发育和生物学功能研究具有重要的作用[11-12].DNA条形码技术在中国襀翅目研究中的前景十分广泛,不仅可以用于物种的快速鉴定,还能促进种群遗传学、生物地理学以及生物多样性等方面的研究. 文中统计和分析了中国襀翅目DNA条形码的序列特征,并验证了该技术对物种区分的有效性. 在未来的研究中需要对中国襀翅目各科、属、种选取大量样本进行DNA条形码测序,并分析不同分类阶元的DNA条形码划分阈值,构建中国襀翅目DNA条形码数据库.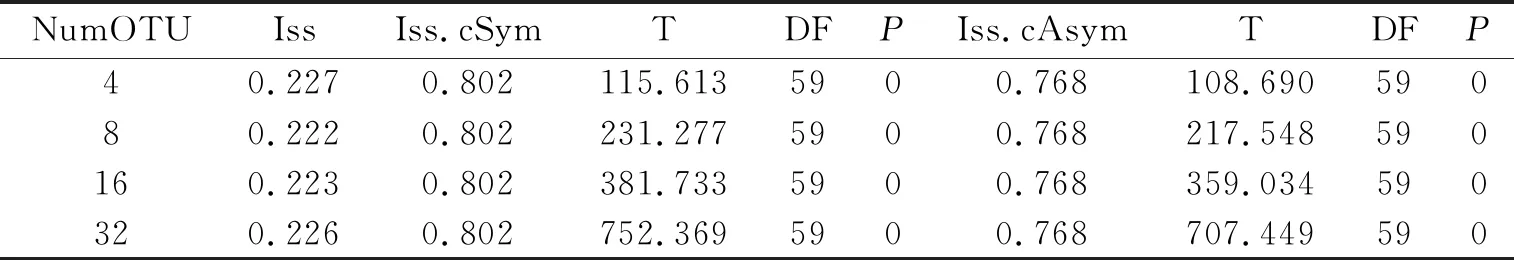
2.3 遗传距离分析
2.4 DNA条形码系统进化树

3 结论
