中年男性,腰痛无力4年,进行性下肢无力3年
——GNE肌病
2021-06-15汪露露朱敏洪道俊
汪露露 朱敏 洪道俊
1 资料
患者,男,31岁,装修工人,因“腰痛无力4年,进行性下肢无力3年”于2020年8月17日就诊于我院。
患者于2016年8月左右感觉腰部无力及疼痛,表现为弯腰吃力,搬重物时起身困难,疼痛在腰部活动后加重,但当时未予以重视。2017年出现左下肢无力,行走时脚尖着地,上抬费力,当时无肢体萎缩,右侧下肢不受累。2018年感腰部无力较前加重,行腰椎MRI提示腰椎间盘突出,保守治疗后无好转。2019年出现右下肢无力,脚尖着地,并发现双下肢,特别是足部肌肉萎缩。2020年感肢体无力较前加重,双上肢受累,表现为双上肢举物困难,上下楼梯困难,并发现双手肌萎缩。病程中双小腿出现胀痛,无吞咽困难,无饮水呛咳,无胸闷及呼吸困难,无肌肉跳动,无复视,无肢体感觉异常。自发病以来,无大小便功能障碍。
既往史和家族史:否认高血压及糖尿病等慢性病史。自幼按计划进行免疫接种,无特殊药物史,无毒物接触史。父母体健,非近亲结婚,有1个哥哥和妹妹,无与患者类似疾病,无其他家族性遗传病。
体格检查:体温36.2℃,脉搏87次/min,呼吸14次/min,血压122 mmHg/87 mmHg,心律齐,叩诊心界不大,各瓣膜区听诊未闻及杂音,其他内科查体未见明显异常。神志清楚,言语清晰流利,记忆力、计算力、定向力、判断力及理解力等高级皮层功能检查未见明显异常。双侧瞳孔等大等圆,直径约2.0 mm,对光反射灵敏。双眼睑闭合力可,双侧眼球向各个方向运动正常,未及复视和眼球震颤。角膜反射正常,双侧面部痛温觉无减退。双侧颞肌、咀嚼肌对称、有力,双侧额纹、鼻唇沟对称。悬雍垂居中,双侧软腭上抬有力,咽反射存在。双侧转颈、耸肩对称有力。伸舌居中。双手骨间肌,双侧下肢肌肉,特别是双侧胫前肌萎缩。颈屈肌力4级,双上肢肌力5上级级,双侧髂腰肌力3级,双股四头肌力 5上级级,腘绳肌力 4级,左踝跖屈肌力 4上级级,左踝跖伸肌力4级,右踝跖屈肌力4级左右,右踝跖伸肌力5上级级。四肢及躯干深浅感觉正常。四肢腱反射减低。双侧指鼻试验正常,跟膝胫试验正常。双侧Babinski’s征和Chaddock’s征均阴性。脑膜刺激征阴性。
辅助检查:血、尿、大便常规正常,血常规、肝肾功能、血糖、血脂、电解质、同型半胱氨酸、凝血功能、血沉、降钙素原、风湿四项、抗核抗体、铁蛋白、血清乳酸等均未见明显异常。肌酸激酶:1806 U/L(正常值 43~172 U/L);肌酸激酶心肌同工酶:14.6 U/L(正常值0~18 U/L)。心电图未见明显异常。肝、胆、胰、脾、双肾彩超未见异常。胸片正侧位未见异常。腰椎MRI提示竖脊肌明显脂肪化,腰椎间盘未见明显突出(图1A、B)。下肢MRI显示大腿平面半腱肌和大收肌显著萎缩伴脂肪化,半膜肌、股二头肌长头、股二头肌短头轻微脂肪化,而股四头肌正常(图1C、D);小腿平面右侧(比目鱼肌、腓骨长短肌明显脂肪化,胫前肌轻微脂肪改变),左侧(比目鱼肌、腓骨长短肌、胫骨后肌、踇长屈肌明显脂肪化,胫前肌轻微脂肪改变)(图1E、F)。四肢神经电图检查示左胫神经感觉传导未引出肯定波形,余神经运动、感觉传导未见异常;双侧胫神经H反射未见明显异常。肌电图提示双侧胫骨前肌静息时可见少量异常自发电位,小力时程正常,大力时程病理干扰相。双侧正中神经和胫神经皮肤交感未见明显异常。

图1 患者肌肉核磁改变 腰椎MRI显示脊旁肌明显脂肪化(A,T2加权),无明显水肿(B,T2压脂),腰椎间盘未见明显突出。大腿肌肉MRI显示半腱肌、股二头肌长头、股二头肌短头及大收肌明显脂肪化,股四头肌正常(C,T2加权),无明显水肿改变(D,T2压脂)。小腿肌肉MRI显示比目鱼肌、腓骨长短肌明显脂肪化,胫前肌轻微脂肪浸润(E,T2加权),腓骨长短肌轻微水肿改变(F,T2压脂)。
病理检查:经知情同意,行左胫前肌肌肉活检。苏木精-伊红(HE)染色显示肌束内结缔组织中-重度增生,肌纤维直径大小变异加大,可见成组及散在分布的小圆状及小角状萎缩肌纤维,部分肌纤维肥大变圆,可见部分的肌纤维出现空泡,其空泡内或周边可见嗜碱性颗粒物质沉积,未见肌纤维坏死再生,及炎细胞浸润(图2A)。Gomri改良三色(MGT)染色显示肌纤维内空泡周物质呈紫红色,提示为镶边空泡(图2B)。过碘酸雪夫(PAS)染色未见深染的肌纤维。ORO染色未见脂肪滴增多。NADH及SDH未见深染肌纤维。
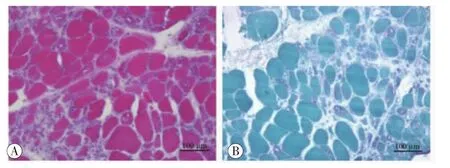
图2 左胫前肌病理改变特点 苏木精-伊红染色显示可见肌束内结缔组组织中-重度增生,肌纤维直径大小变异,可见成组及散在分布的小圆状及小角状萎缩肌纤维,部分肌纤维肥大变圆,可见部分肌纤维出现空泡,其空泡内可见嗜碱性物质沉积,未见炎细胞浸润(A)。Gomri改良三色染色显示空泡肌纤维内物质染色呈紫色,提示为镶边空泡(B)。
基因检查:患者血DNA标本二代测序显示UDP-N-乙酰葡糖胺-2-表异位酶/N-乙酰甘露糖胺激酶(UDPGlcNAc2-epimerase,GNE)基因存在复合杂合突变:分别为c.577C>T,p.R193C和 c.1196C>T,p.P399L, 两个突变均在GNE蛋白的表位酶区,其中c.577C>T在文献中已经报道过[1]。Sanger测序进一步证实该突变的存在,家系验证分析显示c.577C>T来自母亲,c.1196C>T来自父亲,因此符合家系共分离现象。根据临床表现、病理改变特点、基因突变结果,该患者诊断为GNE肌病,也称为Nonaka肌病或伴镶边空泡的远端肌病。
治疗:明确诊断后给予患者N-乙酰神经氨酸(唾液酸)15 g/d,溶于500 mL纯净水中,24 h内分次口服。目前已经治疗4个月,但是患者肢体肌力无明显改善,6分钟步行试验也无明显变化。
2 讨论
GNE肌病是一组由GNE基因突变导致的常染色体隐性遗传性的肌病。在该疾病研究的早期发现该病患者表现突出的肢体远端的肌无力,而肌肉纤维内出现镶边空泡,因此根据临床病理特征曾用名有“远端型肌病伴有镶边空泡”、“Nonaka肌病”、“镶边空泡性肌病而股四头肌不受累”、“遗传性包涵体肌病 2型(IBM2)”等[2]。然而随着致病基因的明确,发现Nonaka肌病和IBM2肌病均为GNE基因突变所致[3-4],而且GNE基因突变导致的临床表现也显著地超出了既往对疾病谱的认识,故目前学界统一用GNE肌病命名[2]。GNE肌病在全世界范围均有报告,发病率约1/100万~9/100万,主要是在伊朗裔犹太人和日本人群中相对常见[5]。
GNE肌病患者发病年龄跨度较大,但通常多在20~40岁,平均28.9岁[1]。典型的临床首发表现为因胫前肌受累而导致足下垂、跨阈样步态,逐渐累及近端、躯干肌肉导致相应的临床症状,少数累及心肌和呼吸肌,一般不累及球部和面部肌肉[5]。病程进展相对缓慢,上肢发病时间平均比下肢症状晚4年,起病到需要轮椅的时间平均11.9年[6]。临床上也有少数患者表现为一些不典型症状,例如以上肢远端无力起病,表现为手握力下降[7];或者类似肢带型肌营养不良发病[8],表现为肢体近端无力为主;或者类似于远端型运动神经病或者长度依赖性运动感觉神经病,表现为周围神经病为主[9];或者以腰部无力、背痛为首发症状,之后出现下肢远端无力[10-11]。该例患者就是以腰部疼痛和无力为首发症状,曾被误诊为腰椎病变而延误确诊时间。
GNE肌病肌肉MRI研究显示:在疾病早期或者不典型临床患者中,股二头肌短头早期即出现显著的脂肪化,伴随臀小肌、胫前肌、拇伸肌、趾长肌、比目鱼肌和腓肠肌内侧头的轻度受累;随着疾病进展,股后部肌群开始受累,且股直肌、股内侧肌和股中间肌也有不同程度的受累,然而股外侧肌在疾病后期仍相对保留正常形态[12-13]。该例患者下肢核磁也表现出类似的受累分布模式,主要是股二头肌短头、大收肌、比目鱼肌受累突出,但是较为特殊的是该例患者的半腱肌也在这个阶段出现显著的脂肪化,是否有一定的特异性需要进一步观察。此外,在疾病早期的腰椎MRI已经显示椎旁肌明显脂肪化,与患者的症状相符,类似肌肉MRI在文献中也有报告[10-11]。
GNE肌病光镜下常表现为慢性肌病样或肌营养不良样的病理改变特点,具体表现为肌内衣的脂肪及纤维结缔组织增生,肌纤维大小不一,肌纤维萎缩,核内移,以及伴随个别肌纤维坏死。比较有特征意义的病理改变为许多肌纤维内出现镶边空泡,然而由于病程和突变位点的不同,不是每个活检都可以看到典型的镶边空泡[1,5]。电镜下在肌纤维的胞浆和细胞核内可出现直径16~18 nm管丝状包涵体,镶边空泡内含大量多层髓样小体,颗粒细丝结构、自噬空泡及细胞碎片等,提示与自噬有关[1,5]。该例患者的肌肉活检表现为典型的GNE肌病的肌肉病理特点,也正是典型的临床改变和病理特点,在获得基因诊断之前,临床上就已经确定了GNE肌病的诊断,后续的基因结果只是验证了我们前期的论断。
GNE基因编码双功能限速酶尿苷二磷酸-N-乙酞氨基葡萄糖2-表位酶/N-乙酰甘露糖胺激酶,该酶具有表位酶和激酶两种功能,是唾液酸生物合成中的限速酶。在GNE肌病中,GNE功能障碍导致的蛋白的唾液酸化障碍,进而介导疾病的过程[5]。目前有大于200个突变在GNE肌病中发现,其中有几个热点突变,Met743Thr在中东,Cys44Ser、Asp207Val、和 Val603Leu 在日本,Val727Met在印度和亚洲[5],而Asp207Val在中国最常见[1]。该例患者两个突变位点均不是热点突变,且有一个位点为文献尚未报道的新突变位点。目前GNE肌病的临床型与基因型的关系尚不明确,有研究指出典型的GNE肌病患者,突变都位于激酶区;具有当有一个突变点位于表位酶区时,患者可能出现不典型的临床症状[14]。然而,另外一项研究显示突变同时在表位酶和激酶结构域上与位于同一结构域的患者相比,临床表现趋于更严重的表型[6]。该例患者的突变点位于表位酶区,因而可以解释腰部起病的不典型临床表现,但是否具有较严重的临床表型还需进一步观察。
由于GNE基因突变后导致唾液酸代谢通路障碍,因此临床上很早就开始关注唾液酸产物替代疗法的研究。早期在动物试验上的研究显示,给GNE突变小鼠补充N-乙酰神经氨酸、乙酰甘露糖胺、6'-唾液乳糖等唾液酸代谢中间产物,均能在一定程度上缓解模型小鼠的及无力症状和病理改变[5]。然而随后在GNE肌病患者的临床观察中,上述代谢中间产物没有对患者起到显著的治疗作用[15]。由于N-乙酰神经氨酸的半衰期非常短,所以该例患者我们采取的事24小时内不定时口服补充治疗,是否有临床效果尚需更长时间随访。
3 点评
肌肉影像学检查包括肌肉超声、肌肉CT、肌肉核磁、肌肉SPECT等检查,这其中肌肉核磁具有多维度、视窗大、软组织分辨率高等优势,已经成为肌肉病变重要的辅助检查手段。肌肉核磁通过分析肌组织的脂肪化程度、肌组织水肿改变、萎缩和肥大肌肉的分布规律、左右分布的对称性等参数,对很多肌肉病变的定位和定性诊断具有较大的参考价值[16]。也可以用于肌肉病变的评估病情进展、治疗效果、运动和康复指导等方面。甚至于对很多肌肉病具有肌肉活检或者基因/抗体检查的等效价值,例如杜兴氏肌营养不良患者的大腿肌肉核磁出现典型的 “三叶一果”征,此种征象的特异性和敏感性都超过95%,在结合典型的临床表型的基础上,临床上几乎可以直接做出诊断[17]。因此,肌肉核磁检查特别适合基层医院对肌肉病的诊断,是对肌肉活检或者分子检查缺如时的一种重要补充。
腰痛在临床上非常常见,引起的病因也较多,常见的原因包括腰背肌筋膜炎、腰椎间盘突出、腰肌劳损等。腰痛也常作为神经系统疾病的首发临床表现,如CADASIL2型和CARASIL腰痛为三联征之一[18],累及中轴的肌病如线粒体肌病、面肩肱肌营养不良、多种酰基辅酶A缺乏症、帕金森病等。腰痛时进行腰椎部位的核磁检查是临床常用到的一种检查,然而临床医生在阅片时关注点通常集中在脊髓及椎间盘上,甚至有时夸大腰椎病变的致病程度,试图去解释患者的腰痛症状。其实腰椎旁肌肉的变性或者无力也是导致腰痛的重要原因,本文这例患者的腰椎核磁就清楚地显示椎旁肌肉的变性和脂肪化。因此,肌肉核磁作为一种相对新出现的检查项目,需要引起各级医生的重视,以提高对肌肉病变的认识和识别度。
