Usher综合征Ⅰ型家系的MYO7A基因变异分析
2021-05-25严提珍黄钧袁德健唐向荣韦笑宝罗世强
严提珍 黄钧 袁德健 唐向荣 韦笑宝 罗世强
作者单位:1 柳州市妇幼保健院医学遗传科 柳州 545001
2 柳州市出生缺陷预防与控制重点实验室 柳州 545001
3 柳州市生殖医学重点实验室 柳州 545001
4 柳州市妇幼保健院耳鼻喉科、听力诊断中心 柳州 545001
5 柳州市妇幼保健院眼科 柳州 545001
Usher综合征(Usher syndrome,USH)是一组以先天性感音神经性耳聋、渐进性视网膜色素变性引起视力障碍、视野缩小为主要临床表现的常染色体隐性遗传性疾病,发病率约为3.3~16.6/100000[1]。依据临床表现的严重程度,可分为I型、II型和III型,其中I型症状最为严重,主要表现为先天性重度或极重度感音神经性耳聋,前庭功能障碍,在青少年时期可引发不同程度的视网膜色素变性[1]。Usher综合征具有高度临床表型和遗传异质性,目前明确可与Usher综合征I型相关的基因有5个(MYO7A、CDH23、USH1C、USH1G、PCDH15)[1],其中MYO7A基因变异发生率最高[2];与Usher综合征II型相关基因有3个(USH2A、GPR98和DFNB31);CLRN1基因与Usher综合征III型相关[1]。临床上Usher综合征主要通过听力检测、眼科检查结合基因检测诊断。本研究通过高通量测序及Sanger测序,发现一个由MYO7A基因c.3631-1G>C和c.6049C>T(p.Q2017*)复合杂合变异导致的Usher综合征I型家系,报告如下。
1 对象与方法
1.1 研究对象
研究对象为先证者及其家系成员,先证者为一名14岁女性,因耳聋和视力异常来我院进行检查,家系成员包括父母、姐姐、妹妹和弟弟(图1)。本研究已获得了柳州市妇幼保健院医学伦理委员会批准,研究对象均签署知情同意书。采集先证者及家系成员的病史(包括出生史、耳聋和眼睛病史、用药史、家族史等),进行系统检查:对先证者及其弟弟进行声导抗、纯音测听、听觉脑干诱发反应(ABR)、畸变产物耳声发射(DPOAE)检查,对其他家系成员进行声导抗和纯音测听;眼科检查包括视力、眼底视野视网膜电图等。
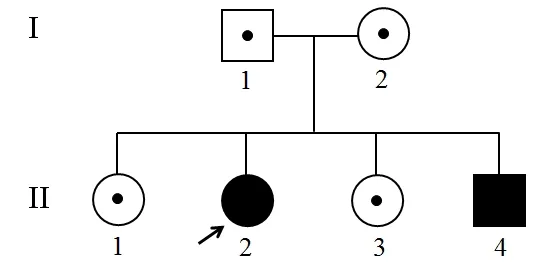
图1 Usher综合征家系系谱图
1.2 方法
1.2.1 样本采集和DNA提取 采集先证者及家系成员EDTA-Na抗凝的外周血4 mL,使用QIAamp DNA Blood Mini Kit提取试剂盒(德国QIAGEN公司)进行基因组DNA提取(操作步骤参照试剂盒说明书)。
1.2.2 医学外显子组测序 用Q800R超声破碎仪将质检合格的基因组DNA打断成片段,打断后DNA片段大小在500 bp以下,峰值约在350 bp。参照NGS文库制备试剂盒(美国Illumina系统)条件及方法进行文库构建,将质检合格的文库稀释至上机测序浓度,完成自动的簇生成及双端测序(美国Illumina HiSeq4000测序仪),通过多种生物信息学软件对测序数据进行分析。结合家系患者的临床表型,对于筛查出来的潜在变异位点,通过检索相关数据库(如HGMD、PubMed、ESP、ClinVar、ExAC库和千人数据等)来判断变异位点的致病性。根据美国医学遗传学学会(American college of medical genetics,ACMG)遗传变异分类标准与指南[3],对变异位点进行评估和致病性分类。
1.2.3 Sanger测序验证变异位点 针对候选变异位点引物,c.3631-1G>C变异位点引物序列为F:5’-CTGTGAAGGGGTATGGGGAG-3’和R:5’-TGAGGGTAGGTGTCTGAGGA-3’;c.6049C >T 变异位点引物序列为F :5’-TGACACACAGAAACCCCTTC-3’和R:5’-TACAAGGATCAGGGGTTGGG-3’。对先证者及其家系成员的DNA样本进行PCR扩增,PCR扩增产物用1.5%琼脂糖凝胶电泳进行鉴定,纯化定量后,送上海立菲生物技术有限公司进行测序检测(美国ABI 3730xl DNA测序仪),测序结果用DNAStar软件的SeqMan与野生型进行序列比对。
1.2.4 氨基酸保守性分析 通过在线软件ClustalW2(https://www.ebi.ac.uk/Tools/msa/clustalw2/)对6个物种目标氨基酸序列进行保守性比对分析。氨基酸序列包括:NP_000251.3(Homo sapiens,人)、NP_001243010.1(Mus musculus,小鼠)、XP_006229806.1(Rattus norvegicus,褐家鼠)、XP_038285928.1(Canis lupus familiaris,犬)、XP_011717460.1(Macaca nemestrina,猕猴)、XP_016777155.2(Pan troglodytes,黑猩猩)。
2 结果
2.1 病史及全身体格检查结果
该家系符合常染色体隐性遗传的特征(图1),先证者(II-2)听力检查结果发现ABR>100 dB HL,而耳声发射不能引出(排除听神经病),双耳纯音听阈均>100 dB HL,为极重度感音神经性耳聋,伴有前庭功能减退(运动发育迟缓,患儿坐立均迟于正常同龄儿,1岁会坐,2岁5个月会走路,缺乏运动技能,不能跑步骑车,冷热水试验无反应)。眼科检查结果显示视网膜电图无反应,b波消失,提示为视网膜色素变性(图2),依据先证者典型的临床表现,临床初步诊断为Usher综合征I型。先证者弟弟(II-4)检查结果也表现为极重度感音神经性耳聋,但还表现为牙齿发育不良、两边眼球大小不一致(左眼球偏小)、斜视、斗鸡眼,无视网膜变性病变。先证者父母(I-1和I-2)、姐姐(II-1)和妹妹(II-3)未出现与先证者类似表型。
2.2 先证者及其他家系成员的测序结果
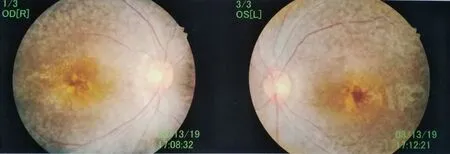
图2 先证者双眼视网膜色素变性
高通量测序检测和分析结果提示,先证者及其弟弟为MYO7A基因c.3631-1G>C和c.6049C>T(p.Q2017*)复合杂合变异(图3 II-2和II-4),通过拷贝数和SNP分析,没有检测到与临床表现可能相关的拷贝数变异。经过Sanger测序验证显示这两个变异位点分别来自先证者的父母(图3 I-1和I-2),先证者姐姐(图3 II-1)和妹妹(图3 II-3)均携带MYO7A c.3631-1G>C杂合变异。
2.3 致病性及保守性分析
c.6049C>T变异位点第2017位氨基酸谷氨酰胺在人、小鼠、褐家鼠、犬、猕猴、黑猩猩多个物种之间高度保守(见图4)。
3 讨论

图3 USH综合征家系Sanger测序结果

图4 保守性分析结果
MYO7A基因相关疾病为常显遗传性耳聋11型、常隐遗传性耳2型和Usher综合征1型[4,5],是常染色体显性/隐性遗传。常显遗传性耳聋11型临床特征主要为语后渐进性听力损失;常隐遗传性耳聋2型临床特征主要为语前和语后重度听力损失;Usher综合征I型临床特征主要为先天性重度或极重度感音神经性听力损失,青春期前视网膜色素变性,伴或不伴前庭功能异常[1]。本例先证者及其弟弟有共同的临床表型,均表现为极重度感音神经性耳聋、前庭功能障碍,但临床表型存在差异,先证者还表现为视网膜色素变性,而其弟弟6岁,无视网膜色素变性病变(可能与年龄有关,后续需密切随访其视力和眼底检查情况),同时具有牙齿发育不良、两边眼球大小不一致(左眼球偏小)、斜视、斗鸡眼等临床表型,这些额外的临床症状在Usher综合征病例中未见报道,提示可能存在其他基因变异或拷贝数变异。该家系两个患者基因型相同,但临床表型有所不同,提示Usher 综合征具有高度表型异质性。
MYO7A基因位于染色体11q13.5区域,含49个外显子,编码2215个氨基酸,编码蛋白属于肌球蛋白家族(myosin 7A),该蛋白为马达蛋白,具有ATP酶活性,可利用水解ATP产生的动能,与肌动蛋白结合,沿着肌动蛋白丝运动[1,5]。该蛋白结构域有3个重要区域:①氨基末端(即马达区域,是肌球蛋白的核心功能区,可与肌动蛋白结合,主要功能是水解ATP和产生动能);②调节区(即颈部,可与肌球蛋白轻链结合形成异亮氨酸-谷氨酰胺基序);③羧基末端:(即尾部,分为SH3结构域、两个FERM结构域和两个MyTH4结构域,主要功能是调节MYO7A运动)。目前已报道超过500个MYO7A基因变异位点可导致Usher综合征I型或遗传性耳聋,变异类型覆盖无义变异、错义变异、移码变异、微缺失变异及剪接变异等[6],分布于各结构域。王圣然等[7]报道了一个由MYO7A基因c.462C>A和EX46_47 Del复合杂合变异导致常染色体隐性遗传非综合征耳聋的家系,其中c.462C>A位于氨基末端,无义变异使得翻译提前终止,产生截短蛋白;EX46_47 Del变异导致了MYO7A羧基末端MyTH4-FERM结构域缺失。刘梦婷等[8]和林长亮等[9]报道的家系分别为c.3412C>T (p.Q1138*)/c.4152G>C(p.K1384N)和c.2694+2T>G/c.6028G>A复合杂合变异,这些变异可引发MYO7A基因编码蛋白质的结构和功能异常,从而导致下游信号转导异常或中断,最终患者出现Usher综合征I型临床症状。
本研究通过高通量测序和Sanger测序基因检测发现先证者及其弟弟均携带MYO7A基因c.3631-1G>C和c.6049C>T(p.Q2017*)复合杂合变异。MYO7A基因c.3631-1G>C为剪接变异,没有在相关临床病例中被报道过,变异位于mRNA剪接区域,序列高度保守,多种计算机辅助算法预测变异可能会影响蛋白功能。依据ACMG变异分类指南[3],该变异符合PVS1(当一个疾病的致病机制为功能丧失的无功能变异)+PM2(千人数据库、ESP数据库、EXAC数据库中正常对照人群中未发现的变异)+PM3(对于隐性遗传基因,在患者反式位置检测到致病或可能致病变异),可判定为致病。MYO7A基因c.6049C>T (p.Q2017*)变异在耳聋相关临床病例中被报道过[10]。该变异为罕见无义变异,无义变异使得翻译提前终止,产生截短蛋白。该变异符合PVS1(同前)+PM2(同前)+PM3(PMID:23767834[10]),可判定为致病。
综上所述,本研究发现了一个MYO7A基因c.3631-1G>C和c.6049C>T复合杂合变异导致Usher综合征I型的家系。c.3631-1G>C新变异丰富了MYO7A基因突变谱。本研究为该家系患者明确了临床诊断,为后续的遗传咨询、产前诊断或植入前遗传学诊断提供了依据。
