Townes-Brocks综合征基因型-表型相关性分析
2021-05-25严晓虹袁慧军赵宇
严晓虹 袁慧军 赵宇
作者单位:1 四川大学华西医院耳鼻咽喉头颈外科 成都 610041
2 四川大学华西医院罕见病研究院 成都 610041
3 陆军军医大学第一附属医院医学遗传中心 重庆 400038
Townes-Brocks综合征(townes-brocks syndrome,TBS)是一种罕见的常染色体显性遗传病。1972年由Townes和Brocks首次描述[1],目前全球已报道超过100个TBS病例,其中我国共报道8例[2~5],这些病例表现出TBS在临床表型上高度的异质性。TBS患者的主要临床特征为肛门闭锁、耳部发育不良(如小耳畸形、耳低位、外耳道闭锁和招风耳等),以及拇指畸形(如轴前多指、拇指三指节畸形和拇指纵裂等)[6]。
SALL1基因是导致TBS的主要致病基因[7]。虽然SALL1基因敲除的小鼠没有表现出类似人类TBS的表型,然而SALL1单等位基因缺失的患者可表现出典型的TBS表型,说明单倍剂量不足是SALL1导致TBS的机制之一[8,9]。同时,存在SALL1突变如p.Arg276X可以逃避无义介导的衰变(nonsense-mediated decay,NMD),产生截断的蛋白质并扰乱正常SALL1蛋白的定位和功能,从而通过显性负效应的机制致病[10]。Miller等[8]发现TBS病人症状的严重程度与SALL1是否通过单倍剂量不足的机制致病没有明显相关性,需要更多的病例、更完整的表型评估和基因数据来完善TBS的基因型-表型相关性的研究。
本文回顾了1998年~2021年1月所有报道携带SALL1变异的TBS病例,以归纳总结TBS表型谱和SALL1变异谱,对SALL1变异导致的TBS进行基因型-表型相关性分析,为后续TBS相关研究提供参考。
1 SALL1基因变异导致TBS的机制研究现状
SALL1是对器官发生至关重要的进化保守基因SALL家族的4个成员之一,其编码的锌指蛋白转录因子与组蛋白脱乙酰酶复合物NuRD相互作用发挥转录抑制功能,并通过SALL1的磷酸化修饰整个过程[11,12]。
SALL1在小鼠胚胎的肾脏、中枢神经系统、肢芽和心脏发育过程中表达,发挥重要作用。SALL1纯合敲除的小鼠通常因出现严重的肾脏发育不良而死亡,而杂合敲除的小鼠没有明显的异常表型[9,13]。SALL1在小鼠肾单位祖细胞和肾基质祖细胞中表达,抑制肾单位祖细胞的分化、维持其干性并以非细胞自主方式限制肾单位祖细胞的过度扩增,通过在后肾表达调节输尿管芽的正常分支[7,9,14~16]。Exner等[17]设计了缺乏SALL基因的非洲爪蟾模型,发现SALL1和Sall4通过抑制Pou5f3家族的表达,以协调神经系统的发育。中枢神经系统对于SALL1的丢失尤为敏感,经典敲除和条件敲除小鼠研究均表明,SALL1影响皮质板层的时间特异性。在没有SALL1的情况下,在胚胎第18.5天(E18.5)大脑皮层的表面积和深度均减少[15]。基因表达谱已鉴定出SALL1是神经系统中小胶质细胞的特征基因(signature gene),对于小胶质细胞的成熟、形态调节和特异性的维持尤为重要[13,18,19]。除了在中枢神经系统中的作用,SALL1还在早期发育阶段的晶状体中高度表达,维持纤维细胞和晶状体的上皮粘附[20]。Morita等[21]发现SALL1可在未分化的心脏祖细胞(cardiac progenitor cells,CPC)中表达,从而驱动左右心室的发生,认为SALL1标记未分化状态的CPC,并调控心脏分化的过程。
在人类中,SALL1的杂合突变可导致TBS,这是一种常染色体显性发育异常,其经典临床三联征为肛门、拇指和外耳的畸形,且常伴随其他系统异常,常涉及肾脏和心脏[22,23]。在全球报道的所有病例中,SALL1变异涉及拷贝数变异(copy number variation,CNV)、移码突变(frameshift variation)、无义突变(nonsense variation)和剪接变异(splicing variation)。
SALL1导致TBS的机制主要分为显性负效应和单倍剂量不足两种。由于绝大多数的SALL1变异导致终止密码子的提前出现,最开始人们推测致病机制应为单倍剂量不足。然而,SALL1杂合敲除的小鼠却没有表现出类似TBS的表型[9]。Kiefer等[24]建立了携带杂合SALL1-ΔZn2-10突变的小鼠模型,该突变导致SALL1缺乏编码大部分锌指结构域的序列,与导致人类TBS的SALL1变异相似。SALL1-ΔZn2-10小鼠表现出了感音神经性聋、多囊肾和四肢骨骼发育异常。另外,翻译出的截断蛋白还可干扰其他Sall家族蛋白的正常功能的行使。2008年,Kiefer等[10]发现SALL1-ΔZn2-10小鼠中可导致发育中的心脏和四肢中两个SALL1下游基因Nppa和Shox2的异位激活,并证明SALL1突变c.826C>T可以产生稳定的截断mRNA和蛋白质(p.R276X),因此认为SALL1主要通过显性负效应导致人类TBS。Bozal-Basterra等[25,26]进一步证明了SALL1p.Pro332Hisfs*10和p.Arg276*导致的截短蛋白可以在人体细胞中的稳定表达,并通过分离培养TBS患者(TBSp.Pro332Hisfs*10和TBSp.Arg276*)的皮肤成纤维细胞和邻近蛋白质组学的检测,发现了截断的SALL1蛋白可以通过影响初级纤毛调节蛋白CCP110、CEP97的正常功能和增加LUZP1的降解,导致细胞中初级纤毛形态和生长频率的异常。研究人员在TBSp.Pro332Hisfs*10成纤维细胞中发现了SHH通路的异常,推测TBS患者的听力异常可能是由于SHH通路异常所导致的。Yang等[27]发现SALL1基因的杂合和纯合敲除小鼠以及携带SALL1-ΔZn2-10的小鼠耳蜗长度变短和外毛细胞数量减少,首次在TBS小鼠中发现了耳蜗的异常发育。
从1999年开始,随着携带SALL1基因CNV的TBS病人的报道(包括SALL1等位基因的缺失),人们意识到单倍剂量不足有可能是TBS的致病机制之一,并通过有限的病例的表型分析推测此类病人的表型比显性负效应导致的TBS的表型轻微[8,28~33]。Miller等[8]通过文献回顾和基因型-表型分析发现上述差异并不成立,存在SALL1拷贝数变异导致的表型严重的TBS患者。
目前只有极少的SALL1蛋白经过实验证明可以产生稳定的截断蛋白[25],因此其余变异是否逃逸了NMD而产生蛋白,或无法表达而导致SALL1单倍体不足,仍然需要进一步研究验证。
2 TBS基因型-表型相关性分析
笔者回顾了1998年~2021年1月所有报道了TBS患者SALL1基因变异情况的文献,希望了解不同位置和不同类型变异导致的TBS临床表型异质性和严重程度的相关性。根据人类基因组变异学会(the Human Genome Variation Society)指南,根据参考序列NM_002968.3以规定的格式进行总结。本文回顾共鉴定156个病例,携带71个不同的SALL1变异,并根据变异类型和所在的锌指结构域进行了划分总结。
图1,2总结了患者的基因型,SALL1的变异类型和所占比例,包括拷贝数变异、移码突变、无义突变和剪接变异。移码突变和无义突变占据了绝大部分,且无一例外全部导致SALL1基因转录的提前终止,3例携带SALL1剪接变异的患者均为内含子内的突变,同样导致转录的提前终止。在10例携带拷贝数变异的患者中,出现了5例SALL1的单等位基因缺失,3例SALL1附近的片段缺失和1例移码突变和框内大片段缺失的复合杂合病例。此外,本文还总结了2例SALL1纯合突变(c.3160C>T纯合突变)。

图1 156例TBS患者基因型总结

图2 156例TBS患者变异位点数量及类型总结
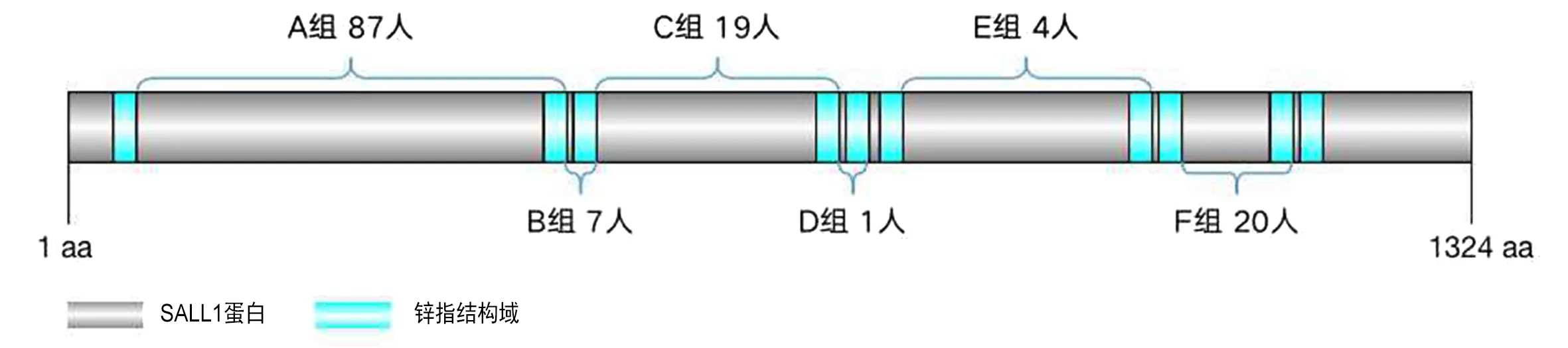
图3 突变位置对应组别及人数

表1 携带SALL1 致病性变异的TBS 患者概况
另外,还存在3 例嵌合突变的情况[34~36],包括2 例染色体核型正常的病例和8 号染色体三体型的病例。核型正常的患者中,其中1例(c.1255delT,p.Leu419Cysfs*74)症状较轻微,文献仅记录患者表现为手部和足部的发育异常,未报道外耳、听力、肛门、泌尿生殖系统及心血管系统异常等TBS常见临床表型,另1例(c.1112C>G,p.Ser371*)患者更为严重,表现为手部畸形、外耳畸形、感音神经性聋、生长发育迟缓和视力发育异常等情况。8号染色体三体型的病例[34]同样表现出典型的TBS表型,伴尿路狭窄、肾功能异常和智力发育迟缓等症状,患者出现了口周、肛周的溃疡,类似症状在其他TBS病例中未报道[35]。
2.1 SALL1的移码突变和无义突变位置与TBS表型严重程度相关
本文根据TBS患者携带的变异类型及变异对应的氨基酸位置进行了分组和表型总结,变异对应的氨基酸位置处于同一个锌指转录结构域的变异分为同一组(图3),表型分类方式参考J.Kohlhase[6]发表在GeneReviews的文章《Townes-Brocks Syndrome》,将TBS表型分为主要表型、次要表型和其他表型,表1~4总结了患者的分组情况及各组在主要表型和次要表型上的阳性率。
主要表型包括①肛门畸形;②外耳发育异常;③手部畸形。次要表型包括①听力损失;②足部畸形;③泌尿生殖系统畸形;④先天性心脏病。除以上表型以外的临床表型纳入其他表型范围。
笔者发现大多数患者的突变(87/136,64.0%)位于A组第65~471位氨基酸之间,这个区间的突变导致SALL1基因只保留了1个完整的锌指结构域[37]。该组病人中TBS典型表型的阳性率很高,外耳、肛门和手部畸形的阳性率分别达到了87.06%,76.47%和83.53%,此外,次要表型如听力损失、足部畸形和泌尿生殖系统异常的阳性率也过半。另外两组样本量较多的C组和F组的病人,突变后SALL1基因分别保留了3个和8个完整的锌指结构域。C组的病例的典型表型的阳性率都与A组病人相似,但是F组的阳性率要明显低于A、C两组。笔者推测,由于F组保留的锌指结构域基序更多,能够更大程度上维持蛋白质的正常功能,因此患者症状更轻微。Netzer等[38]的研究显示,SALL1基因有两个关键的转录抑制结构域,分别位于蛋白质的最N端87个氨基酸区域和第434~690个氨基酸之间,失去两区域中任意一个或破坏第434~690个氨基酸区域的完整性会明显降低SALL1的抑制活性,删除第690个氨基酸之后的区域则对抑制活性的影响不大,该研究结果与A、C组的患者表型较F组严重的情况一致,A、C组的患者存在SALL1关键转录抑制结构域对应编码区域的破坏,而F组的患者突变位置所对应的蛋白质区域位于关键转录抑制结构域之后。
虽然B、D、E组的患者人数过少,难以进行比较,但从以上结果推测,对于SALL1移码突变和无义突变导致TBS的患者,突变位置与表型严重程度具有相关性,位于关键转录抑制结构域对应的编码区域的突变对SALL1蛋白转录抑制活性的影响更大,患者的表型更严重。

表2 携带SALL1 致病性变异的TBS 患者主要表型总结
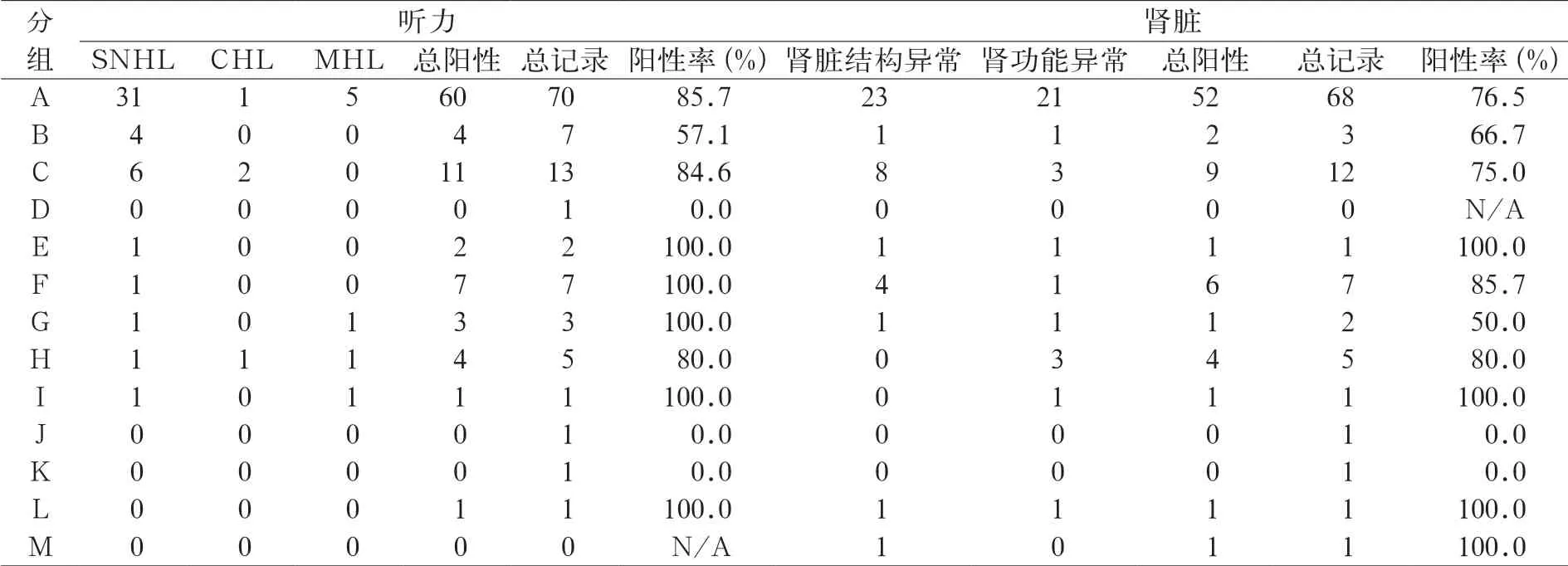
表3 携带SALL1 致病性变异的TBS 患者次要表型听力和肾脏总结

表4 携带SALL1 致病性变异的TBS 患者次要表型心脏、下肢和眼总结
2.2 SALL1拷贝数变异
H-M组患者携带SALL1或SALL1邻近区域的拷贝数变异。H组9个病例存在SALL1基因单等位基因缺失,TBS的典型表型和次要表型的阳性率都很高,且有5/9个病例存在发育迟缓。Borozdin等[39]报道了5例存在SALL1拷贝数变异的病人,认为SALL1缺失患者的TBS表型似乎比其他患者更轻微,后经Miller等[8]对后续发表病例进行进一步补充报道和总结认为Borozdin等人的观点缺少充分证据支持。I组为Borozdin等[39]报道的1例SALL1单等位基因部分片段缺失的病人(g.4785579_4788962del),该CNV导致SALL1的所有双锌指结构域对应的编码序列缺失,根据Netzer等[38]的研究结果,该病例丢失了一个关键的转录抑制结构域。病人表现出典型TBS三联征和多系统的异常,包括肾功能损害、膀胱输尿管返流、足部畸形和眼底异常等。J组的唯一患者携带了5号和16号染色体结构变异t(5;16)(p15.3;q12.1),不清楚断裂位置是否包含或直接破坏了SALL1的连续性,该患者除TBS三联征以外,文章没有报道其他系统的异常。K组和L组的3例患者都只存在SALL1的3’或5’端UTR区的片段缺失,基因编码区本身保留了完整性,但是患者仍表现出典型TBS三联征,提示缺失的区域可能包含重要的调控序列,对基因的功能有关键调控作用。
M组的患者携带了一个SALL1 2号外显子内1.1 kb的片段缺失(c.1488_2049del和p.Gln497Argfs*12),其中只有6 bp的片段保留了下来。Southern blot analysis发现该变异导致了2号外显子内Hind Ⅲ限制性酶切位点(Hind Ⅲ restriction site)的丢失,并产生了另外的4 kb谱带。该患者表现出TBS三联征、发育迟缓以及肾脏发育不良[33]。
2.3 剪接变异总结
Blanck等[40]报道了一个SALL1IVS2-19T>A突变导致TBS的家系。该家系的3个患者没有表现出完整的TBS三联征,即肛门、拇指和外耳的畸形、先证者合并有尿道下裂、肾脏发育不良、肾功能衰竭和房间隔缺损,另外两个携带同样突变的亲属都没有其他系统的异常。
该报道通过RT-PCR证明了变异产生了与野生型SALL1等量的转录本,对RT-PCR的片段测序显示突变SALL1的转录本出现异常剪接而插入17 bp序列,导致SALL1蛋白过早终止(1208 aa)。说明SALL1致病性变异不止存在于外显子中,内含子的变异也可导致终止密码子的提前出现,从而导致TBS。提示对于存在TBS表型的患者,如果全外显子测序未查见SALL1致病性变异,应考虑该基因内含子区域是否存在变异。
2.4 TBS存在常染色体隐形遗传的情况
Vodopiutz等[41]报道了目前唯一一个TBS常染色体隐性遗传的家系。先证者及其妹妹携带SALL1c.3160C>T纯合变异,表型包括法洛四联症、多囊肾、围产期发生的慢性肾功能衰竭、肢体及外耳畸形、感音神经性聋、胼胝体发育不良、皮质性盲和精神运动功能完全不发育(complete lack of psychomotor development),研究人员将这种表型称为中枢神经系统TBS(central nervous system-townes-brocks syndrome,CNS-TBS)。该家系中11人携带SALL1c.3160C>T杂合变异,除了患者父亲存在无需干预的室间隔缺损外,家族其余成员均健康,无肛门闭锁、耳聋或肾功能异常的家族史。由于并非家族内所有杂合变异的携带者都接受了全面的临床检测,且不能确定先证者父亲的心脏结构异常是否由SALL1杂合变异导致,因此笔者认为,尚不能确定CNS-TBS的遗传模式是常染色体隐性遗传,有可能是CNS-TBS的不完全外显导致。同时Vodopiutz等也发现SALL1c.3160C>T(p.Arg1054*)突变能产生少量的截断蛋白,可能保留了部分功能,可解释为什么SALL1c.3160C>T杂合突变的携带者没有明显的表型,该推测与Netzer等[38]的研究结果一致。
F组患者突变位点都位于SALL1c.3160C后,且表型明显,只是相较于突变位置靠前的患者稍轻微。因此,至少对于位置在关键转录抑制区域对应编码区以外的变异,不能完全根据突变位置或位点的数量推测症状的严重程度。
3 结论
笔者对目前报道的存在SALL1变异的TBS患者进行了回顾和基因型-表型相关性分析,初步得出以下观点:①位于SALL1关键转录抑制结构域对应的编码区域的突变与其他位置的突变导致的TBS相比,前者的患者的表型更严重;②突变位于关键转录抑制结构域对应的编码区域之后的SALL1基因可能维持部分功能,此类杂合突变携带者可不出现明显TBS表型;③至少对于位置在关键转录抑制区域对应编码区以外的变异,不能完全根据突变位置或位点的数量推测症状的严重程度。
SALL1变异可以通过单倍剂量不足或显性负效应两种机制导致TBS,然而,对绝大部分因SALL1变异导致终止密码子提前出现的TBS患者而言,其携带的变异基因是否能产生稳定的截断蛋白依然不清楚。对这类患者,其表型可能是通过上述两种机制中任意一种造成,因此,仅靠变异位点的位置进行基因型-表型分析并不全面。
未来,需要建立大样本的TBS患者队列,通过多组学(如转录组、蛋白组和代谢组等)结合的方法全面地研究患者体内不同的SALL1基因变异的表达情况和与健康对照在分子层面的差异,才能进一步阐明不同基因型与表型之间的相关性,并探索相关分子网络的变化,以明确SALL1变异导致TBS的致病机制。
