CLPP基因新变异导致Perrault综合征的致病性鉴定
2021-05-25周凯卢宇袁慧军瞿申红陆秋天黄兰诚林钻平唐凤珠
周凯 卢宇 袁慧军 瞿申红 陆秋天 黄兰诚 林钻平,5 唐凤珠
作者单位:1 广西壮族自治区人民医院耳鼻咽喉头颈科 南宁 530021
2 广西医科大学研究生院 南宁 530021
3 陆军军医大学第一附属医院医学遗传中心 重庆 400038
4 四川大学华西医院罕见病研究院 成都 610041
5 桂林医学院研究生院 桂林 541004
Perrault综合征(perrault syndrome,PRLTS)是一种罕见的常染色体隐性遗传性疾病,其特征是女性卵巢发育不良和男/女性感音神经性耳聋(sensorineural hearing loss,SNHL)[1]。PRLTS的临床表现具有高度的异质性,还可能合并共济失调、神经系统疾病、小头畸形、癫痫、生长发育迟缓和智力障碍等表现。PRLTS根据致病基因不同可以分为6种亚型,分别由HSD17B4、HARS2、CLPP、LARS2、TWNK、ERAL1基因突变所导致[2~4],其中CLPP基因突变所导致的疾病称为PRLTS 3型[1]。迄今为止,CLPP基因已有14个致病性突变被报道[5~11],国内尚未见CLPP基因突变导致PRLTS的相关报道。本研究通过耳聋基因靶向测序在一个临床诊断为PRLTS家系的2名患者中检测到CLPP基因的两个新发变异,明确了该家系致病原因,为多系统症状的诊治提供了指导。
1 材料与方法
1.1 家系资料
本家系的研究经过陆军军医大学第一附属医院伦理委员会审批,由患儿监护人签署知情同意书。对家系成员进行详细问询,包括耳聋相关病史、肾病家族史、既往史、耳毒性药物史、女性月经史以及外伤史等。
1.2 方法
1.2.1 听力学检查及表型鉴定 先证者及家系成员进行听力学检查,包括纯音测听和声导抗检查等。对患者进行常规查体、听力检查和耳科专科检查等。根据家系调查和听力学结果绘制系谱图。
1.2.2 耳聋基因靶向测序 采用由袁慧军团队研发的耳聋基因大规模平行测序方法对受检对象的158个耳聋致病基因进行检测,其中包括已知非综合征型耳聋基因和常见综合征型耳聋基因。用安捷伦公司定制目标区域捕获试剂盒(agilent technologies,santa clara,CA,USA),根据试剂盒说明书 进行基因组DNA建库,捕获158个基因的所有外显子、侧翼区内含子和剪接区域。精确定量后,在Illumina HiSeq 2000分析仪上对捕获的DNA片段进行测序。按照标准Illumina程序进行数据分析和生物信息学处理。使用BWA软件包(http://bio-bwa.sourceforge)将测序所得短片段序列与GRCh37/hg19参考基因组数据集比对,识别可能影响蛋白质功能的非同义变异,候选变异通过公共数据库和内部外显子组数据库中等位基因频率小于0.005进行筛选。对新发现罕见变异的患者,收集家庭成员信息和血样,通过Sanger测序对候选变异进行基因分型及共分离分析。
1.2.3 变异验证 全部家系成员的验证:参考人类基因组序列数据库(GRCh37)针对CLPP基因检出的变异设计引物序列(见表1),PCR扩增反应条件:a.95℃预变性2分钟,b.95℃变性30秒,退火温度62~58℃,c.每个循环降0.5℃,退火30秒;d.72℃延伸30秒,扩增30个循环后,e.72℃再延伸2分钟,最后10℃保存,扩增后的产物进行sanger测序分析。
2 结果
2.1 临床表现及听力结果
该家系共2代5人(1人已故,4人在世),Ⅱ-2为女性,现年16岁,纯音测听提示双耳平均听阈大于100 dB HL,表现为双耳极重度NSHL(见图1),且有明显的智力障碍表现,合并四肢肌无力,平衡功能及乳房发育差,月经初潮年龄推迟(目前尚未月经初潮)等生殖系统发育异常表现。Ⅱ-3为男性,现年14岁,双耳平均听阈大于100 dB HL,表现为双耳极重度NSHL(见图2),合并癫痫,平衡功能差,智力及生殖系统发育未见明显异常,头颅MRI提示脑白质病变,双侧小脑沟增宽,脑电图结果为成人中度异常脑电地形图,基本节律慢化,痫电发放。其父母(I-1,I-2)听力均正常,智力及生殖系统未见明显异常,家系分析符合常染色体隐性遗传特征(见图3),患儿父母否认近亲婚配,且无耳聋、智力障碍家族史。
2.2 耳聋基因检测结果
耳聋基因大规模平行测序结果表明,2名患儿均为CLPP基因c.661G>T和c.328delA复合杂合变异。
2.3 Sanger测序验证及家系内验证
家系成员Sanger测序结果与高通量测序结果一致,2名患儿CLPP基因c.661G>T和c.328delA位点的复合杂合变异,患儿父母分别携带CLPP基因c.661 G>T和c.328 delA位点的杂合变异(见图4、5)。
2.4 变异致病性分析
在gnomAD人群数据库未见CLPP基因c.661G>T和c.328delA变异报道,经查询自建数据库中7202例中国听力正常成年人的外显子组数据,为新的罕见变异。CLPP基因c.661G>T(p.Glu221Ter)和c.328delA(p.Ser110AlafsTer17)分别为无义变异和移码变异,根据ACMG变异解读标准将CLPP基因c.661G>T和c.328delA突变位点均判定为致病性变异。
3 讨论
本研究通过高通量耳聋基因靶向测序,在一个PRLTS家系中发现CLPP基因c.661G>T和c.328delA复合杂合突变致病,为本研究新发现的两个致病性变异。该家系中男性患者表现为耳聋合并癫痫,平衡功能差,但智力及生殖系统发育未见异常,女性患者表现为耳聋合并智力障碍,四肢肌无力,平衡功能差以及乳房发育差、月经初潮年龄推迟(目前尚未月经初潮)等生殖系统发育异常表现。与PRLTS 3型描述的表型相一致[1]。
CLPP基因包含6个外显子,编码的ClpP是一种ATP依赖性蛋白酶,通过N端靶向序列定向到线粒体基质[12~14]。Kang等[15]通过电子显微镜确定纯化的重组人ClpP和ClpX分别形成七聚体和六聚体环,2个ClpP七聚体环和2个ClpX六聚体环组成ClpXP复合体,分解异常折叠的蛋白质。ClpX决定复合物的底物特异性,展开异常蛋白并将其送入ClpXP复合体形成的腔中,在ATP存在下分解蛋白,平均产物长度为10个残基,这些产物被外肽酶进一步降解为游离氨基酸。

表1 Sanger 测序引物

图1 Ⅱ-2纯音测听结果
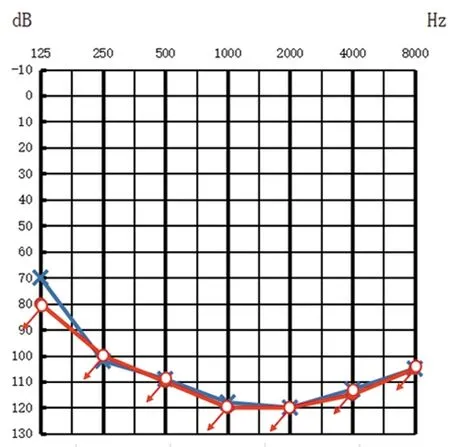
图2 Ⅱ-3纯音测听结果
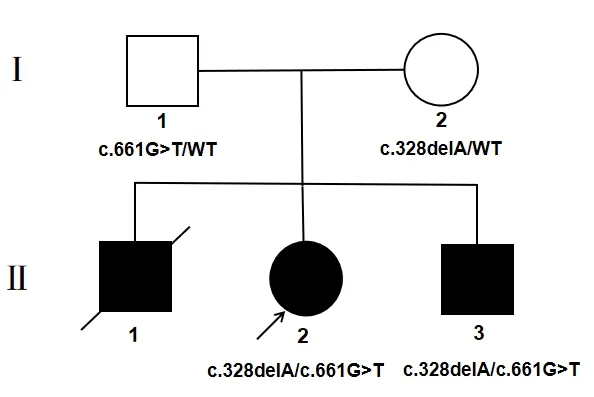
图3 PRLTS家系图:基因型与表型共分离

图4 c.661G>T野生型(WT)和突变型(Mut)反向Sanger测序验证峰图

图5 c.328delA野生型(WT)和突变型(Mut)反向Sanger测序验证峰图
CLPP基因突变改变ClpXP复合体形成的桶形腔的结构,导致不稳定的ClpP,其半衰期缩短,细胞ClpP水平降低,损害异常折叠的线粒体蛋白的分解,线粒体蛋白稳态异常,导致线粒体功能障碍[12,16],对CLPP基因高表达的组织器官影响明显,这些变化是如何导致Perrault综合征尚不明确。Gispert等[16]通过基因敲除获得了Clpp-/-小鼠,存活的基因敲除鼠和同窝野生型小鼠在哺乳期体重相仿,此后其体重增加较慢,成年后基因敲除鼠的体重小于野生型小鼠。雌、雄的Clpp-/-小鼠完全不育,表现出自发的运动活动减少、耳聋,并且比野生型更早地自然死亡。
Jenkinson等[17]首次报道了一个来自英国-巴基斯坦的近亲家庭(PDF1)的3名患PRLTS的姐妹,在先天性极重度SNHL的基础上均表现出不同程度的卵巢发育异常。Jenkinson等[6]通过连锁分析、纯合子定位和外显子测序,确定了该家系的致病原因为CLPP基因第4号外显子c.433A>C(p.thr145pro)纯合突变,该突变发生在头部区域中第1个β折叠的β-3链内高度保守的残基处。该突变与疾病在家系中共分离,在193个同族对照和NHLBI Exome Variant Server (ESP6500)数据库中均未发现。除了严重的先天性感音神经性听力丧失和卵巢早衰外,3个姐妹还表现出身材矮小、小头畸形、癫痫发作、中等程度的学习困难以及小腿和小脑共济失调并伴有下肢痉挛等症状。这些表现与本研究报道的先证者表型高度重合。
CLPP基因迄今为止已有10余个与耳聋相关的突变位点被报道。本研究报道了2个PRLTS患者CLPP基因两个新的变异c.661G>T和c.328delA,经sanger测序验证以上两个突变分别来自患者父亲和母亲,且父母双方听力正常,无其他综合征表现,符合常染色体隐性遗传模式,在gnomAD等人群数据库中未见报道。CLPP基因c.328delA(p.Ser110AlafsTer17)为移码突变,引起翻译蛋白质截断。c.661G>T(p.Glu221Ter)为无义突变,使得肽链合成提前终止,肽链长度变短。两种截断突变均有可能引发无义介导的mRNA降解,从而没有蛋白质产物。根据ACMG变异解读标准将CLPP基因的两个新发变异位点c.661G>T和c.328delA突变位点均判定为致病变异。
PRLTS男性患者可能仅表现为耳聋,但女性患者则常合并卵巢发育不良。本研究通过基因诊断明确了该家系的耳聋患者为PRLTS,该家系中两名患者年龄较小,女性患者除耳聋表型外,还存在智力障碍,运动功能差等表现,在女性特征方面,患者目前双侧乳房发育差,且尚未出现月经初潮,这提示卵巢功能发育不良可能性大,需提示父母注意相关表型,及时到专科就诊。男性患者除耳聋表型外,合并癫痫发作,运动、平衡功能发育较差,表现为走路不稳、易摔跤、无法完成跳绳、跳远等动作。
综上所述,在中国PRLTS家系中发现CLPP基因的两个新发变异位点c.661G>T和c.328delA为尚未报道的致病变异,该位点的报道扩大了CLPP基因的突变谱,明确了致病原因,为多系统症状的诊治提供了指导,为该家系提供了临床遗传咨询。
