超高效液相色谱-串联质谱法测定沙拉酱中16种喹诺酮类药物残留
2021-05-20黄思聪杨建涛黄象金
◎ 黄思聪,杨建涛,黄象金
(肇庆市食品检验所,广东 肇庆 526040)
喹诺酮类药物是以4-喹诺酮为母核的一类广谱抗生素,因其价格低廉且抗菌谱广,广泛运用于动物及水产养殖[1]。长期摄入喹诺酮类抗生素,会使人体出现过敏反应和变态反应,影响免疫系统功能,产生耐药性,不利于疾病治疗[2-3]。目前,世界卫生组织(WTO)、欧盟及我国都对喹诺酮类抗生素在养殖方面有着严格的使用标准。根据我国农业部公告第278号,禁止在产蛋鸡中使用达氟沙星、培氟沙星、环丙沙星、诺氟沙星、恩诺沙星、氧氟沙星、二氟沙星、沙拉沙星和洛美沙星等各种形式的制剂;颁布于2015年的农业部公告第2292号,则决定对用于食品动物中的洛美沙星、培氟沙星、氧氟沙星和诺氟沙星4种原料药及制剂停止生产、经营及使用。
从过往的监测数据可以得知,鸡蛋、鸡肉类的喹诺酮类抗生素滥用问题依然突出[4-10]。而针对以鸡蛋为重要原材料的沙拉酱,目前并没有有效的检测喹诺酮类抗生素的方法或标准。截至2017年,沙拉酱行业产量约20.2万t,表观消费量约18.9万t[11]。面对如此庞大且不断增长的消费市场,建立一个单独和新型的检测技术或方法显得越来越迫切。
目前,测定喹诺酮类抗生素残留的检测方法有酶联免疫法[12-13]、高效液相色谱法(HPLC)[14-16]、液相色谱-串联质谱法(LC-MS/MS)[17-18]。相对于酶联免疫法具有较高的检出限;HPLC灵敏度低,无法提供目标物的结构信息;LC-MS/MS对目标物的分离能力强、灵敏度更高、操作方便,更适合对低残留多组分的同时测定。前处理方法主要有液液萃取法[19]、QuEChERS[20-22]、固相萃取法[22-26]等。
除鸡蛋外,沙拉酱中的大量色拉油亦是导致影响检测结果的重要因素,因此,选择一种高效准确的前处理方法同样很有必要。本研究运用Captiva EMR-Lipid 固相萃取技术,对沙拉酱中的16种喹诺酮类抗生素进行提取净化,该技术具有回收率和准确率高,分析速度快等优点,进一步联合UPLC-MS/MS法,达到更加高效准确地对沙拉酱中喹诺酮类抗生素残留量的 测定。
1 材料与方法
1.1 材料与试剂
氧氟沙星、恩诺沙星、环丙沙星、达氟沙星、沙拉沙星、双氟沙星、培氟沙星、诺氟沙星、洛美沙星、司帕沙星、伊诺沙星、吡哌酸、萘啶酸、恶喹酸、西诺沙星和氟甲喹标准物质,纯度均大于97%,德国Dr.Ehrenstorfer公司生产;乙腈、甲醇、甲酸,质谱纯,美国Merck公司生产。
1.2 仪器与设备
UPLCH-class/Xevo TQD超高效液相色谱-串联质谱联用仪(美国Waters公司)、XSE205DU电子分析天平(梅特勒-托利多仪器(上海)有限公司)、comfortⅡ超纯水系统(德国Sartorius公司)、MultiReax多试管振荡器(德国heidolph公司)、H1750R高速台式冷冻离心机(湖南湘仪实验室仪器开发有限公司)、EVA-60全自动浓缩仪(美国Reeko公司)及Captiva EMR-Lipid净化柱(美国安捷伦公司)。
1.3 方法
1.3.1 样品前处理
严格称取粉碎混匀后的沙拉酱样品4.0 g,置入 50 mL离心管中,再精确加入80%乙腈水(含5%甲酸)10 mL及一颗陶瓷均质子,涡旋振荡1 min,振荡提取2 min,10 000 r·min-1在4 ℃离心5 min,室温静置;准确移取5.0 mL上清液经Captiva EMR-Lipid净化柱净化,重力自流,轻度负压抽干小柱,收集流出液,用40 ℃氮吹浓缩近干,再加入1.0 mL初始流动相复溶,通过0.22 μm微孔滤膜过滤,最后滤液供UPLC-MS/MS 测定。
1.3.2 溶液配制
标准储备液:准确称取喹诺酮类抗生素标准品各10 mg,分别溶解于甲醇中,并定容至100 mL,最后配制成100 μg·mL-1质量浓度的标准储备液。
1.3.3 液相色谱条件
色谱柱:Waters BEH C18(100 mm×2.1 mm,1.7 μm); 流动相A为0.1%甲酸水,流动相B为甲醇;流速 0.35 mL·min-1;进样量2.0 μL;柱温35 ℃。梯度洗脱 程 序:0~1 min,95%流 动 相A;1~4 min,95%→65%流动相A;4~7 min,65%→5%流动相A;7~7.1 min,5%→95%流动相A;7.1~10 min,95%流动相A。
1.3.4 质谱条件
离子化模式:电喷雾离子源(ESI),正离子扫描;扫描模式:多反应监测(MRM);碰撞气:氩气,纯度>99.999%;脱溶剂气温度:550 ℃;脱溶剂气:氮气,流速1000 L·h-1;毛细管电压:1.0 kV。
1.4 数据处理
每组实验重复6次,结果表示为平均值,采用SPSS 16.0软件进行数据分析。
2 结果与分析
2.1 质谱条件的优化
将浓度为1.0 μg·mL-1的喹诺酮类抗生素单标通过质谱端蠕动泵进样。在正离子模式下,通过一级质谱扫描(MS1 Scan),调节毛细管电压(Capilary)、脱溶剂气流量、脱溶剂气温度以获取稳定的、响应强度最大的[M+H]+分子离子,以确定母离子;通过子离子扫描模式(Daughter Scan)在50~400 m/z范围内找出丰度较强的碎片离子,选择丰度最强的碎片离子为定量离子,优化后的碰撞能量(Collision)、锥孔电压(Cone)等参数如表1所示。
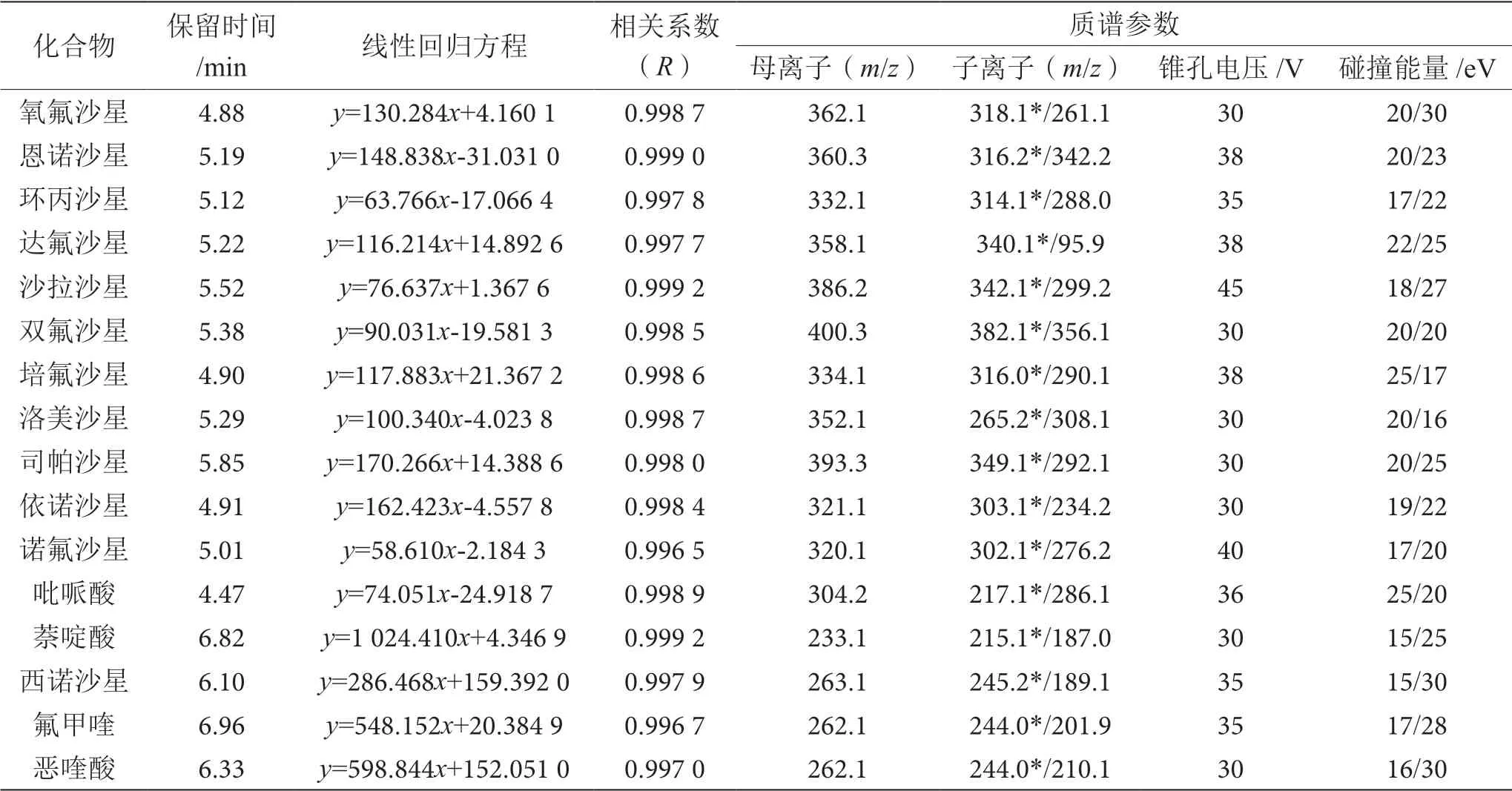
表1 分析物的线性范围、相关系数、保留时间及质谱参数表
2.2 提取溶剂的选择
根据喹诺酮类抗生素的极性,结合Captiva EMRLipid前处理方法,分别选择甲醇、乙腈、酸化乙腈和EDTA-Mcllvaine缓冲溶液作为提取溶剂,比较空白沙拉酱加标回收实验提取效果。结果表明,由于沙拉酱的脂肪含量高达76.2%,乙腈具有较强的提取能力,对喹诺酮类抗生素的提取效果最好,结合Captiva EMRLipid方法可有效去除蛋白质和脂肪;根据喹诺酮类化合物易溶于酸性或碱性溶液的特质,因此在本实验中分别采用1%、2%和5%的甲酸乙腈进行提取,发现当甲酸浓度为5%时,蛋白沉淀效果最好;对5%的甲酸乙腈进一步添加20%的水,样品分散和目标物释放更快更彻底,同时可以更好地活化Captiva EMR-Lipid柱。因而本实验选用80%乙腈水(含5%的甲酸)作为提取溶剂。
2.3 流动相的选择
采 用Waters BEH C18柱(100 mm×2.1 mm, 1.7 μm)作为分析柱,考察甲醇-水、甲醇-0.1%甲酸水、乙腈-水、乙腈-0.1%甲酸水、甲醇-10 mmol·L-1乙酸铵和乙腈-10 mmol·L-1乙酸铵流动相对喹诺酮类抗生素的分离效果及其质谱响应情况。结果表明,在ESI源正离子模式下,流动相中添加甲酸,通过帮助溶液中的正离子质子化,进而产生更强的电喷雾信号,提高目标物质检测的灵敏度,且峰形良好,故选用0.1%甲酸水作为水相;另外,使用甲醇相对乙腈,分离效果和响应强度更佳。综合分析,采用甲醇-0.1%甲酸水为流动相时,喹诺酮类抗生素的质谱响应值较高,且经梯度洗脱后能够有效分离(见图1)。因此选择甲醇-0.1%甲酸水作为流动相。
2.4 样品净化方法的选择
使用QuEChERS-EMR-Lipid(以下简称Q-EL)和Captica EMR-Lipid(以下简称C-EL)两种提取净化方式分别对浓度为1.0 μg·kg-1、20.0 μg·kg-1、 100.0 μg·kg-1的3组加标样品进行测定,每个添加水平测定6次,对结果进行比较。结果表明,Q-EL法的加标回收率为47.5%~103.0%,RSD值为1.1%~10.9%;C-EL法的加标回收率为72.5%~104.0%,RSD为1.3%~7.5%。这是因为在净化过程中,Q-EL法是先对目标物中的脂质进行选择性萃取,去除多余的水分从而改善对目标物的分配,但由于其除脂作用依靠的是无水MgSO4,MgSO4遇水会结块,影响环丙沙星、培氟沙星、依诺沙星、诺氟沙星和吡哌酸这几种药物的净化效果,导致回收率偏低;而C-EL法由于采取体积排阻和疏水相相互作用捕获脂类,将体积较大的分析物排阻出去,可以最大限度地减少离子抑制对目标分析物的影响。
因而相对Q-EL法,使用C-EL法的可靠性和稳定性更高,可以达到更好的净化效果,回收率更高,精密度更理想。不同的处理方法平均回收率和精密度见表2。
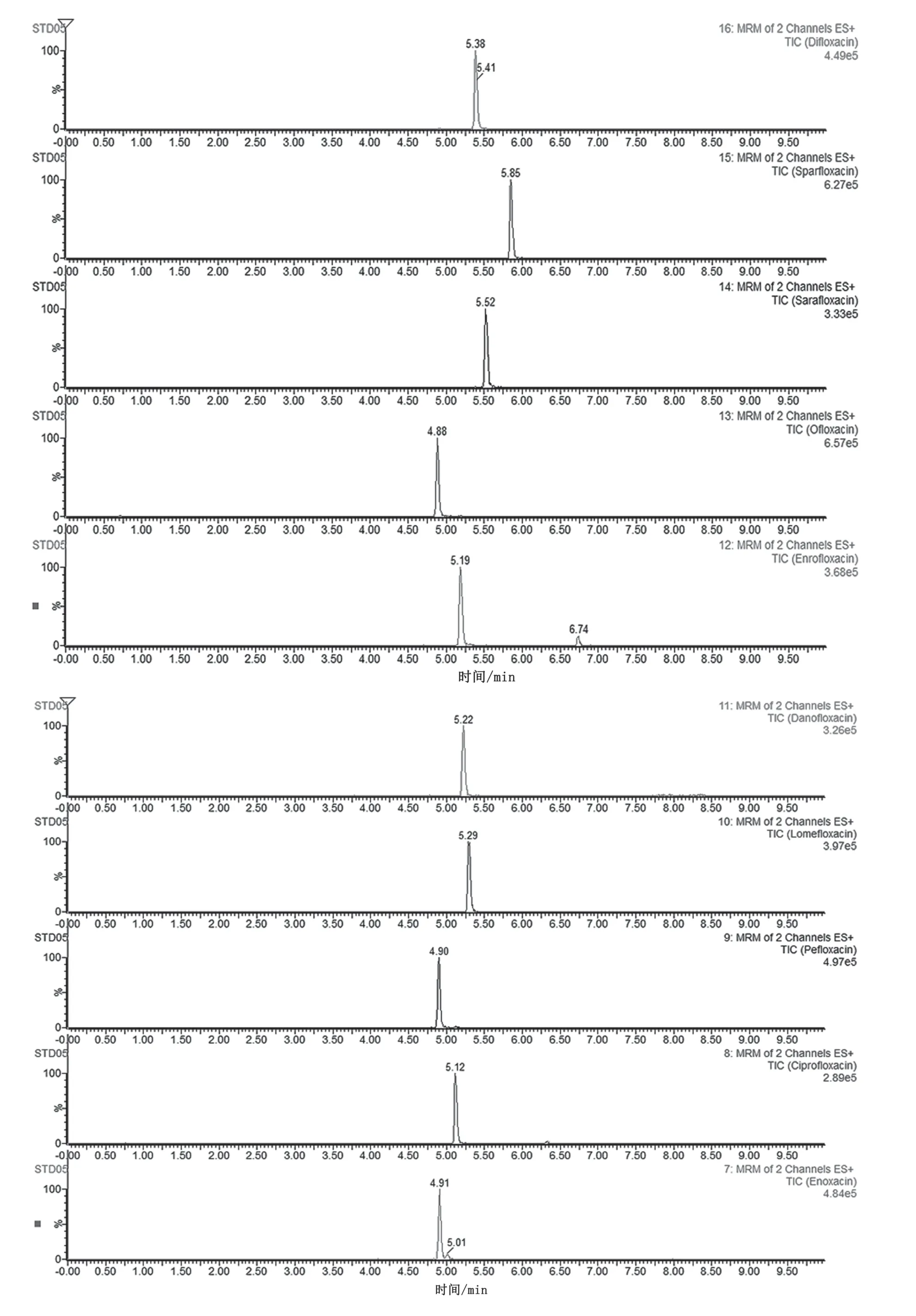

图1 喹诺酮类抗生素提取离子总离子流图
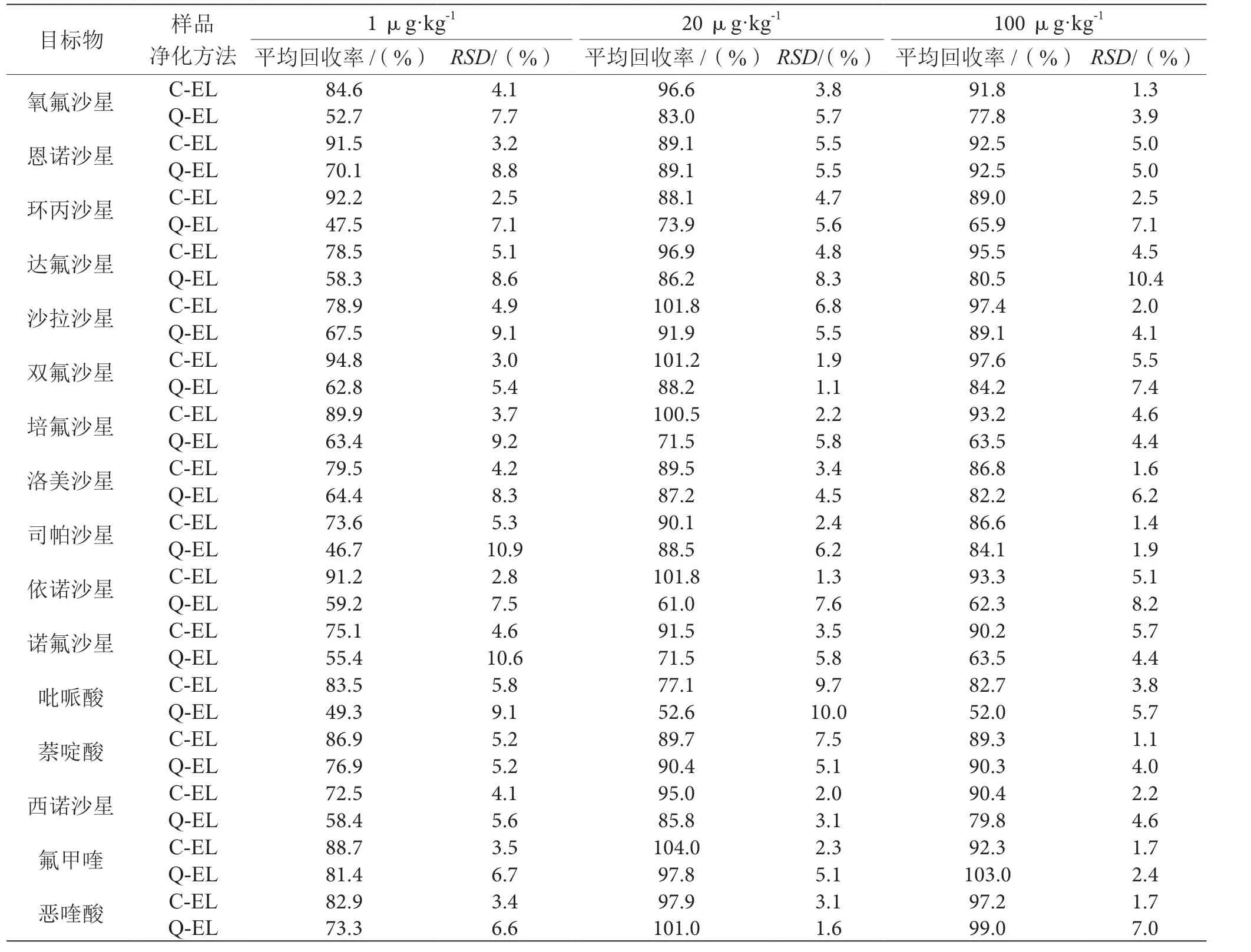
表2 不同的处理方法平均回收率和精密度表(n=6)
2.5 基质效应的消除
采用UPLC-MS/MS分析样品时,基质效应是影响定量结果准确性的重要因素。因而为消除 UPLC-MS/MS法测定沙拉酱基质中喹诺酮类抗生素的基质效应,采用标准添加法,分别对纯溶剂以及不含目标物的空白沙拉酱样品的基质效应进行分析,按式(1)计算基质效应ME值。

式(1)中,Sm为空白基质匹配标准曲线的斜率,Ss为纯溶剂标准曲线的斜率。若ME>0,表现为基质增强效应;若ME<0,表现为基质抑制效应。基质效应测定结果见表3。
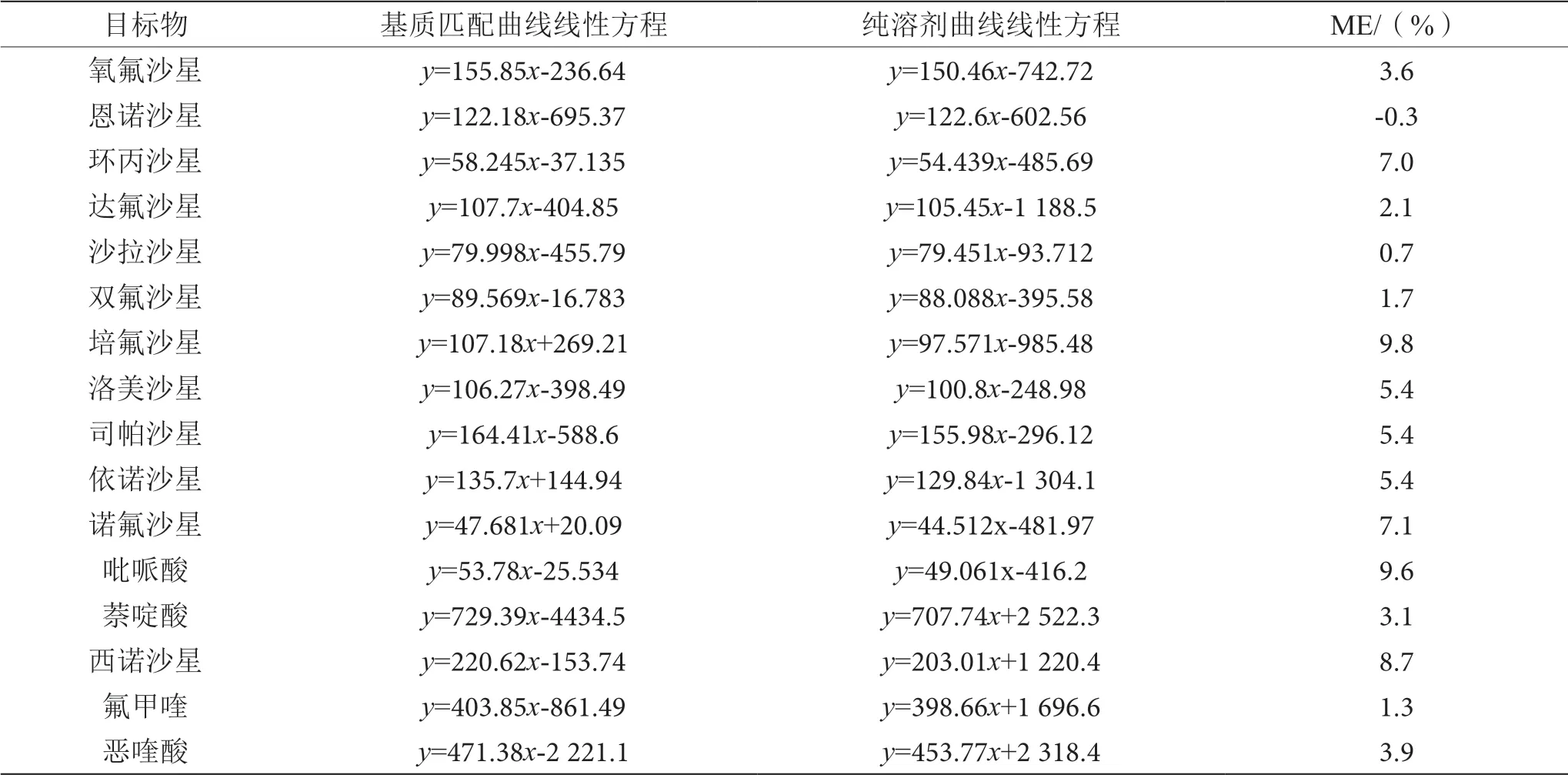
表3 基质效应表(n=6)
由表3可知,在纯溶剂和不含目标物的空白沙拉酱样品两种基质中分别加入1 μg·kg-1、10 μg·kg-1、25 μg·kg-1、50 μg·kg-1、100 μg·kg-1和200 μg·kg-1的喹诺酮类抗生素标准溶液,按上述Captica EMRLipid固相萃取-UPLC-MS/MS法测定目标物质谱响应强度,重复测定6次,取6次平均响应值分别制得基质匹配曲线和溶剂标曲线,得出ME值为-0.3%~9.8%,表明在0.1~200.0 μg·kg-1质量浓度范围内,采用Captica EMR-Lipid固相萃取-UPLCMS/MS法测定沙拉酱中喹诺酮类抗生素时存在基质干扰,表现为基质增强效应。本实验采用基质匹配方法消除基质效应影响,对16种化合物均采用基质匹配外标校正的方法,以确保实验结果的准确性。
2.6 线性范围、检出限与定量限
按 照0.5 ng·mL-1、1.0 ng·mL-1、5.0 ng·mL-1、 10.0 ng·mL-1、20.0 ng·mL-1、50.0 ng·mL-1、100.0 ng·mL-1和200.0 ng·mL-1共8个质量浓度对喹诺酮类抗生素标准溶液进行配制,使用UPLC-MS/MS法测定,横坐标(x)表示目标物质量浓度,纵坐标(y)表示质谱响应值,据此绘制标准曲线。以信噪比S/N=3结合空白基质噪音响应确定方法的检出限(LOD)为 0.5 μg·kg-1,以信噪比S/N=10确定方法的定量限(LOQ)为2.0 μg·kg-1。由表1可知,在0.5~200.0 ng·mL-1范围内,喹诺酮类抗生素质量浓度与其质谱响应值呈良好的线性关系,相关系数均大于0.996 5。
2.7 加标回收率和精密度
对不含目标物的空白沙拉酱样品,依次添加 1.0 μg·kg-1、20.0 μg·kg-1、100.0 μg·kg-1的16种喹诺酮类抗生素标准溶液,同样使用上述UPLC-MS/MS法对每个添加水平进行6次测定,进一步计算回收率和相对标准偏差(RSD),由表2可知,在1.0~100.0 μg·kg-1加标范围内,喹诺酮类抗生素的平均加标回收率为72.5%~104.0%,RSD为1.3%~7.5%,测定结果达到方法学的要求,表明该方法具有较高的准确度和良好的精密度。
2.8 实际样品分析
按本研究所建立的Captica EMR-Lipid固相萃取-UPLC-MS/MS法测定20份市售沙拉酱中喹诺酮类抗生素残留量,均未检出氧氟沙星、恩诺沙星等16种喹诺酮类抗生素的残留。
3 结论
本研究采用Captica EMR-Lipid技术提取净化沙拉酱中的氧氟沙星、恩诺沙星、环丙沙星、达氟沙星、沙拉沙星、双氟沙星、培氟沙星、诺氟沙星、洛美沙星、司帕沙星、依诺沙星、吡哌酸、萘啶酸、恶喹酸、西诺沙星和氟甲喹,成功建立UPLC-MS/MS法同时测定沙拉酱中喹诺酮类抗生素的方法。该方法前处理过程简单、灵敏度和准确度高、分离效果好,可为沙拉酱中喹诺酮类抗生素残留检测及确证提供新的方向;同时对进一步加强我国食品检测技术能力,弥补该领域食品检测技术的空白及保障食品安全具有十分重大的意义。
