酸铝胁迫土壤中耐铝大豆根际不同部位细菌群落结构、功能及其对促生菌富集作用的研究
2021-05-19文钟灵杨旻恺陈星雨郝晨宇任然储淑娟韩洪苇林红燕陆桂华戚金亮杨永华
文钟灵,杨旻恺,陈星雨,郝晨宇,任然,储淑娟,韩洪苇,林红燕,陆桂华,戚金亮,杨永华
酸铝胁迫土壤中耐铝大豆根际不同部位细菌群落结构、功能及其对促生菌富集作用的研究
文钟灵,杨旻恺,陈星雨,郝晨宇,任然,储淑娟,韩洪苇,林红燕,陆桂华,戚金亮,杨永华
南京大学植物分子生物学研究所,医药生物技术国家重点实验室,生命科学学院,南京 210023
针对酸性土壤中影响作物生产的主要限制因子(pH及其铝毒),选用耐酸铝且具有固氮能力的豆科作物是改良该类土壤、促进农业生产的有效措施之一,至于其所关联的根际微生物是否起到相应的促进作用,一直为国内外学者所关注和探究。为此,本研究以铝耐受型大豆品种基因型(BX10)和铝敏感型大豆品种基因型(BD2)为材料,以酸性红壤为生长介质,采样部位按照土层到根系的距离由远到近的顺序划分为:根外对照土(bulk soil, BS)、两侧根际土(rhizospheric soil at two sides, SRH)、刷后根际土(rhizospheric soil after brush, BRH)和冲洗后的根际土(rhizospheric soil after wash, WRH)。利用Illumina MiSeq对16S rRNA基因扩增产物的高变区V4进行高通量测序,研究了不同耐铝基因型大豆根际细菌群落的结构、功能与分子遗传多样性的差异性作用。结果表明,各处理间大豆根际细菌群落的alpha多样性无显著性差异,beta多样性差异也均不显著。PCA和PCoA分析可见BRH和WRH部位的物种组成较为一致,而BS和SRH部位具有相似的物种组成,说明植物生长主要影响根际的BRH及WRH部位的微生物,对SRH影响较小。对各分类水平物种组成和丰度进行比较,门分类水平三元图表明两个基因型大豆均在WRH部位富集蓝细菌门(Cyanobacteria)细菌;统计分析表明铝耐受型大豆(BX10)根部对于增强植物抗逆性的植物根际促生菌(plant growth promoting rhizobacteria, PGPR)有富集作用,这些富集的细菌包括蓝细菌门、拟杆菌门(Bacteroidetes)和变形菌门(Proteobacteria)等,以及部分与固氮和耐铝的功能相关的属种。另对同一个基因型大豆不同采样部位间进行比较分析,结果显示土壤不同采样部位可以选择性富集不同的PGPR物种。此外,16S rDNA的同源蛋白簇(clusters of orthologous groups of proteins, COG)功能预测分析的结果表明,多个COG包括COG0347、COG1348、COG1433、COG2710、COG3870、COG4656、COG5420、COG5456和COG5554均可能与固氮直接相关;BD2相比于BX10,结果显示在BRH和WRH部位似乎均更易富集固氮直接相关的COG,其可能的原因尚待进一步研究。
酸性土壤;耐铝大豆;根际;细菌群落;植物根际促生菌
在粮食和土地与人口增长之间的矛盾日益尖锐的今天,传统的耕作栽培技术已经根本无法满足人们的需求,为了促进农业生产,选用何种适宜的农作物一直是国内外所关注的问题。大豆是世界上重要的蛋白质和油源,在酸性土壤中大量栽培。由于大豆的生物固氮能力,其可以作为优良的轮作和间作作物,以提高土壤肥力和结构[1]。全球可耕地的50%为酸性土壤,在中国酸性土壤约占土地总面积的20%[2]。大多数酸性土壤中,植物生长受到以下几个因素的限制,包括铝的毒性水平,以及一些必需元素的如氮(N)、磷(P)和一些微量营养素的不足,导致作物产量低[3]。氮元素是植株生长必需的大量营养元素,自然界的氮主要以氮气形式存在,无法被植株直接利用[4],豆科植物与根瘤菌之间所形成的共生固氮体系具有较强的共生固氮能力,这种能力使豆科植物成为自然界氮输入的重要来源,同时也成为人类食物中蛋白质的重要来源[5]。铝是酸性土壤作物生产的主要限制因子之一(pH<5)[6],酸性土壤条件下,铝以有活性的毒性的形式释放到土壤溶液中,达到能抑制根系生长或损伤根的水平,进而抑制水分和矿物质的吸收,降低根系活力,降低叶片光合作用,抑制大豆生长[7,8]。植物对铝毒害的抗性机制很多,如有机物的渗出、根际pH的变化、细胞壁的交换性结合等[9]。植物可以释放有机酸、酚类化合物、多肽和其他化合物作为根系分泌物,减少酸性土壤中的铝毒害。有机酸能螯合有毒铝元素和活化磷元素,铝毒害和磷缺乏均可激活有机酸的外排[10],大量证据表明根系有机酸分泌是植物耐铝的主要机制[11]。
根际是根的表面和贴近根的周围的土层,一般指离根轴表面数毫米范围之内,是土壤–根系–微生物相互作用的微区域。根际有益微生物,主要是指对植物生长发育具有直接或间接促进作用的或对植物根际有害微生物具有拮抗作用的土壤微生物,其中植物根际促生菌(plant growth promoting rhizobacteria, PGPR)可以改变土壤中无效矿质元素的形态,使其更容易被植物吸收利用[12]。
本研究使用的大豆品种为铝耐受型大豆Baxi 10 (BX10)和铝敏感型大豆Bendi 2 (BD2),BX10大豆起源于巴西,比起源于中国广东的BD2大豆更耐铝[13]。前者在Al3+处理下受到的根系生长抑制作用比后者轻[14]。这两个大豆基因型因其不同的耐铝性而受到广泛的研究。之前的研究表明,与铝敏感型大豆BD2相比,铝耐受型大豆BX10具有更大的柠檬酸盐流出速率和产量,磷脂脂肪酸在不同时期也存在差异。此外,BX10根际土壤中革兰氏阴性菌(GN)和革兰氏阳性菌(GP)的比值与BD2相比存在变化[15,16]。那么,除了植物有机酸分泌这一常见抗酸铝策略外,铝耐受型大豆BX10和铝敏感型大豆BD2的根际会不会通过对于不同微生物的富集(例如各种PGPR),成为BX10在酸性土壤中获得更好生长状态的又一因素呢?因此,本研究通过对土壤根际微生物的16S rDNA(V4可变区)扩增子进行测序分析,试图分析BX10和BD2对根际微生物群落和微生物代谢活动的影响,探讨大豆根际的不同部位与大豆根际微生物群落之间的联系。
1 材料与方法
1.1 植物、土壤材料及取样方法
本研究选择两个基因型的大豆((L.) Merr.):BX10 (Baxi 10,耐铝基因型)和BD2 (Bendi 2,铝敏感基因型)[13]。种植前,我们对大豆种子使用95%乙醇消毒30 s,2.5%次氯酸钠消毒5 min,并使用无菌水冲洗3~5遍。酸性土壤(pH 4.43,交换性铝含量为1.45 cmol/kg)取自中国科学院江西鹰潭红壤生态试验站(28.208° N,116.937° E)[17]。本研究中使用的根际箱的具体规格与本课题组之前报道的一致,专利申请公布号为CN 102175487 A[15,16]。根际箱的具体标准为长200 mm、宽150 mm、深200 mm,在中部种植大豆时,用尼龙薄膜将根际箱分为5个部分,以限制根而不限制水分和营养[18]。本研究在开花期采集土壤样品(种植后60天),采样方法沿用前人的研究并稍作了修改[19,20]。根据采样部位到根系的距离由远到近的土层分为4个部分:根外对照土(bulk soil, BS)、两侧根际土(rhizospheric soil at two sides, SRH)、刷后根际土(rhizospheric soil after brush, BRH)和冲洗后的根际土(rhizospheric soil after wash, WRH):用磷酸盐缓冲盐水冲洗后,在4000×下离心10 min收集)。所有这些样本在–80℃下保存用于提取DNA。
1.2 DNA提取及16S rDNA扩增子测序信息
使用PowerSoil DNA分离试剂盒(MoBio Laboratories Inc, USA)提取DNA,操作流程与先前研究一致[21,22]。提取DNA后在1%琼脂糖凝胶上评估DNA样本的质量,并使用Qubit荧光计(Qubit 2.0, Invitrogen, USA)对样本进行量化,以尽量减少微生物群落调查中的可变性[23]。对提取后基因组DNA进行检测,确保每份DNA样品的浓度皆大于0.4 ng/μL[23]。采用改良后的双端达到250 nt的高通量测序,使用引物515F(5ʹ-GTGCCAGCMGCCGCGGTAA-3ʹ)和806R (5ʹ-GGACTACHVGGGTWTCTAAT-3ʹ)扩增V4区[24,25]。进行PCR扩增和PCR产物纯化的步骤方法如之前文章所述[22,26]。通过华大基因有限公司(中国武汉)在Illumina MiSeq平台上进行高通量测序。共24个测序数据已上传NCBI,SRA编号为PRJNA 613772 (接收后自动释放)。
1.3 alpha多样性及beta多样性分析
本研究使用Rank-Abundance曲线来解释物种丰度/均匀度。采用Venn图和Pan/Core分析方法计算多个样本中共有/共享的OTU数目。根据OTUs和物种注释结果进行alpha和beta多样性分析。为了分析环境中微生物物种多样性的复杂性,本研究采用alpha多样性来反映群落丰富度(sobs指数、Chao指数和Ace指数)、群落多样性(Shannon指数和Simpson指数)和群落覆盖度(Coverage指数),而beta多样性分析采用QIIME (v1.8.0)计算,然后用不同的距离矩阵对样本的物种复杂度差异进行评价和对比[27,28]。本研究使用主成分分析(Principal Component Analysis, PCA)和主坐标分析(Principal Co-ordinates Analysis, PCoA)来研究样本群落组成的相似性或差异性。三元图结果分析参照Bulgarelli等人提出的方法[29]。
1.4 功能预测分析和统计分析
16S rDNA的COG功能预测分析分类通过PICRUSt软件在I-Sanger平台上进行(http://www. i-sanger.com)。相似性分析(the analysis of similarities,ANOSIM)和置换多因素方差分析(permutational multivariate analysis of variance, PERMANOVA,又称Adonis分析)使用I-Sanger平台,利用silva128/16S数据库(http://www.arb-silva.de)根据最小样本数序列对样本进行抽平。在统计分析中,显著性差异主要使用单因素方差分析(one-way ANOVA)。整个16S rDNA测序结果的数据分析参照了刘永鑫等人介绍的方法[30]。
2 结果与分析
2.1 BX10和BD2根际细菌群落的alpha多样性总体无差异
在这项研究中,根际箱的设计和使用与本课题组之前的报道一致[15,16]。24个样品的测序片段总数为892,331,平均每个样品约37,180个片段,范围从36,311~40,950不等。各样本序列平均长度约为253 bp。Rank-Abundance曲线比较光滑,说明24个样品的丰度和均匀度较高,测序深度和质量均满足要求,说明OTU覆盖了根相关细菌群落中足够的可检测物种(附图1)。Pan分析和Core分析发现所有样品共有的物种数为472,而所有样品所含物种总数为3697(附图2)。
为了比较分析BX10和BD2根际细菌群落的alpha多样性,研究首先分析了不同alpha多样性指数的稀释曲线。稀释曲线表明,测序数据量合理,数据量大,覆盖率足够,足以反映绝大多数微生物多样性信息。图1显示了6个不同alpha多样性指数的结果。sobs、Chao和Ace指数(图1,A~C)和覆盖指数(图1F)的结果表明,各样本的群落丰富度和群落覆盖度无显著性差异。虽然Shannon指数(1D)在各样本间无显著差异,Simpson指数(1E)的结果表明,BX_16FWRH的群落多样性低于BD_16FWRH。
2.2 BX10和BD2根际细菌群落beta多样性无显著性差异
Venn图显示,每个样品的OTU数相似,而BX10和BD2中BS、SRH、BRH和WRH的总共享OTU分别为1581和1582 (附图3)。此外,BX_16FSRH、BX_16FBRH、BX_16FWRH、BD_16FWRH、BD_16FBRH和BD_16FWRH共有OTU数目为1437个。通过OTU绘制的PCA和PCoA图,对比检测花期BX10和BD2的BS、SRH、BRH和WRH样品之间OTU组成的差异(图2)。主成分分析PCA图表明,同一技术重复的样品聚为一组,同一取样部位不同基因型大豆之间未显著区分(图2A)。基于Bray Curtis的主坐标分析PCoA图表也得到了相同的结果(图2B),且BX_16FBRH、BX_16FWRH和BD_16FWRH聚集更紧密。基于weighted Unifrac距离的PCoA与基于Bray Curtis的PCoA图表相似,而基于unweighted Unifrac距离的PCoA图显示在BX10和BD2根际细菌群落中,BS和SRH距离较近,而BRH和WRH更可能聚集在一个组中(附图4)。
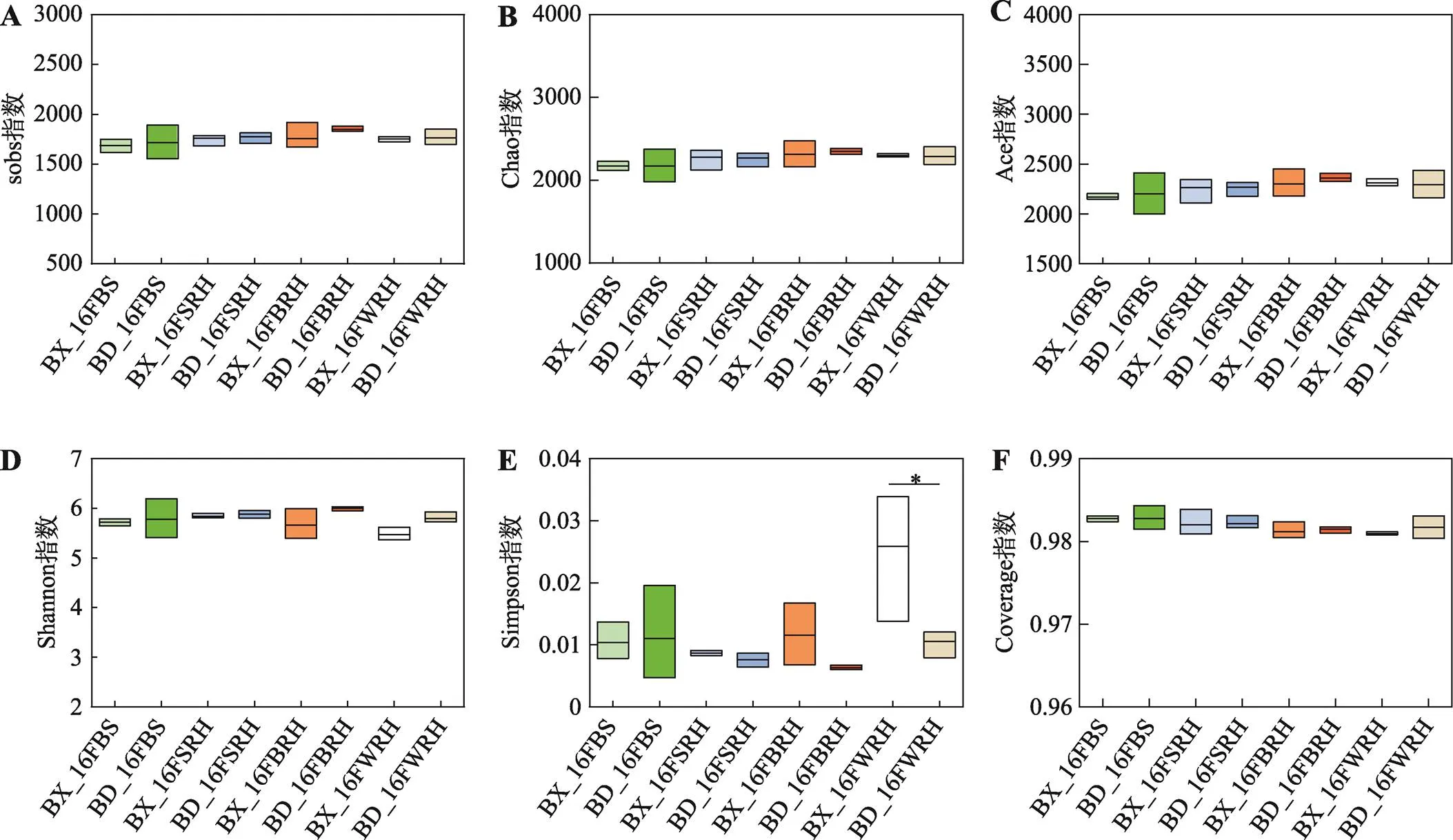
图1 Alpha多样性的箱型图
A:sobs指数的alpha多样性的箱型图;B:Chao指数的alpha多样性的箱型图;C:Ace指数的alpha多样性的箱型图;D:Shannon指数的alpha多样性的箱型图;E:Simpson指数的alpha多样性的箱型图;F:Coverage指数的alpha多样性的箱型图。BX和BD分别代表耐铝大豆BX10和铝敏感大豆BD2;F代表花期;BS、SRH、BRH和WRH分别代表根围土、两侧根际土、刷后根际土和冲洗后根际土;显著性检验采用单因素方差分析,*表示BX10和BD2组之间存在显著差异(<0.05)。
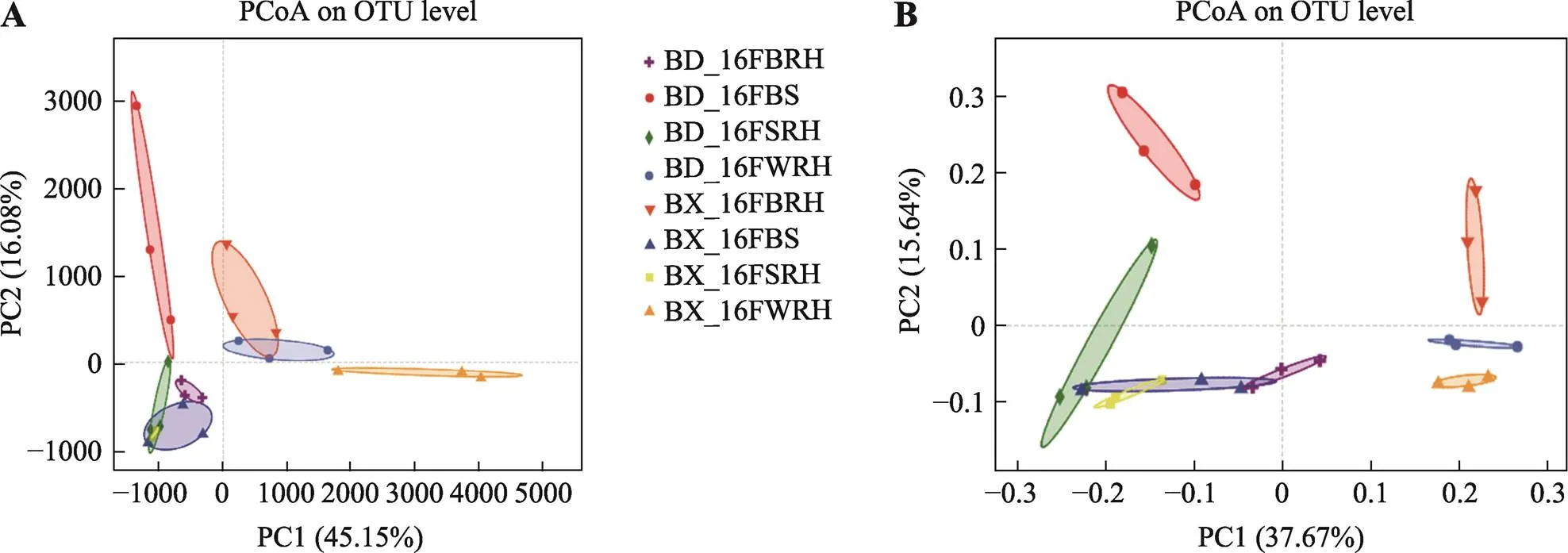
图2 细菌群落丰度的主成分分析和主坐标分析图
A:细菌群落OTU丰度基于欧氏距离的主成分分析图;B:细菌群落OTU丰度基于Bray-Curtis距离的主坐标分析图。详细处理信息同图1。
ANOSIM和Adonis分析的结果表明,同一采样部位,BX10和BD2根际细菌群落的beta多样性在统计学意义上仍无显著差异(>0.05);同时,BX10的4个不同采样部位之间以及BD2的4个不同采样部位之间的根际细菌群落beta多样也均无显著差异(>0.05) (表1)。
2.3 大豆基因型和采样部位影响了根际细菌群落各分类水平的组成
根据样品的分类分析结果,可以直观地了解每个样品在不同的分类层次(门、纲、目、科、属、种)的细菌群落组成和丰度。本研究首先选取了相对丰富最高的13个主要门分类水平物种进行作图比较(图3)。如图所示,丰度最高的6个门分别是变形菌门(Proteobacteria)、酸杆菌门(Acidobacteria)、绿弯菌门(Chloroflexi)、疣微杆菌门(Verrucomicrobia)、拟杆菌门(Bacteroidetes)和放线菌门(Actinobacteria)。并且,蓝细菌门(Cyanobacteria)在不同采样部位差异明显,特别是WRH部位极高(图3)。
以3个根际采样部位绘制的三元图的结果表明,两种基因型大豆的根部微生物群落,都存在蓝细菌门在WRH部位富集的情况(图4)。因为蓝细菌门的丰度在WRH部位的丰度为17.05%和18.92%,其他部位平均丰度仅为3.5%,证明测序结果并未被植物叶绿体严重干扰,故蓝细菌门丰度数据是可信的。
属分类水平物种组成丰度分析表明,各样本之间,根瘤菌属()的丰度无显著性差异。伯克氏菌属()在BX_16FBRH中丰度高于所有其他样品。此外,还存在嗜酸栖热菌属()在BD_16FBS显著高于其他样本,以及粘液细菌属()存在BX_16FBRH>BX_ 16FSRH,BD_16FBRH>BD_16FSRH的现象,即在FBSH这个部位对属存在富集作用(附表1)。
种分类水平物种组成丰度分析可以找到8个种(表2),分别是埃氏慢生根瘤菌(),约氏不动杆菌(),短波单胞菌(),脆假单胞菌()和放射型根瘤菌(),他们属于变形菌门;氧化假节杆菌()和甲基营养节杆菌()属于放线菌门,浅绿气球菌()属于厚壁菌门(Firmicutes)。统计学分析(one-way ANOVA)的结果表明,的丰度在各样品之间无显著性差异,在BD_ 16FWRH中的丰度远高于BD2的其他采样部位。、、和都发现在BD_16FBS的丰度远高于其他样本,而和的丰度则有BX_ 16FBRH大于BX10其他样品的情况。
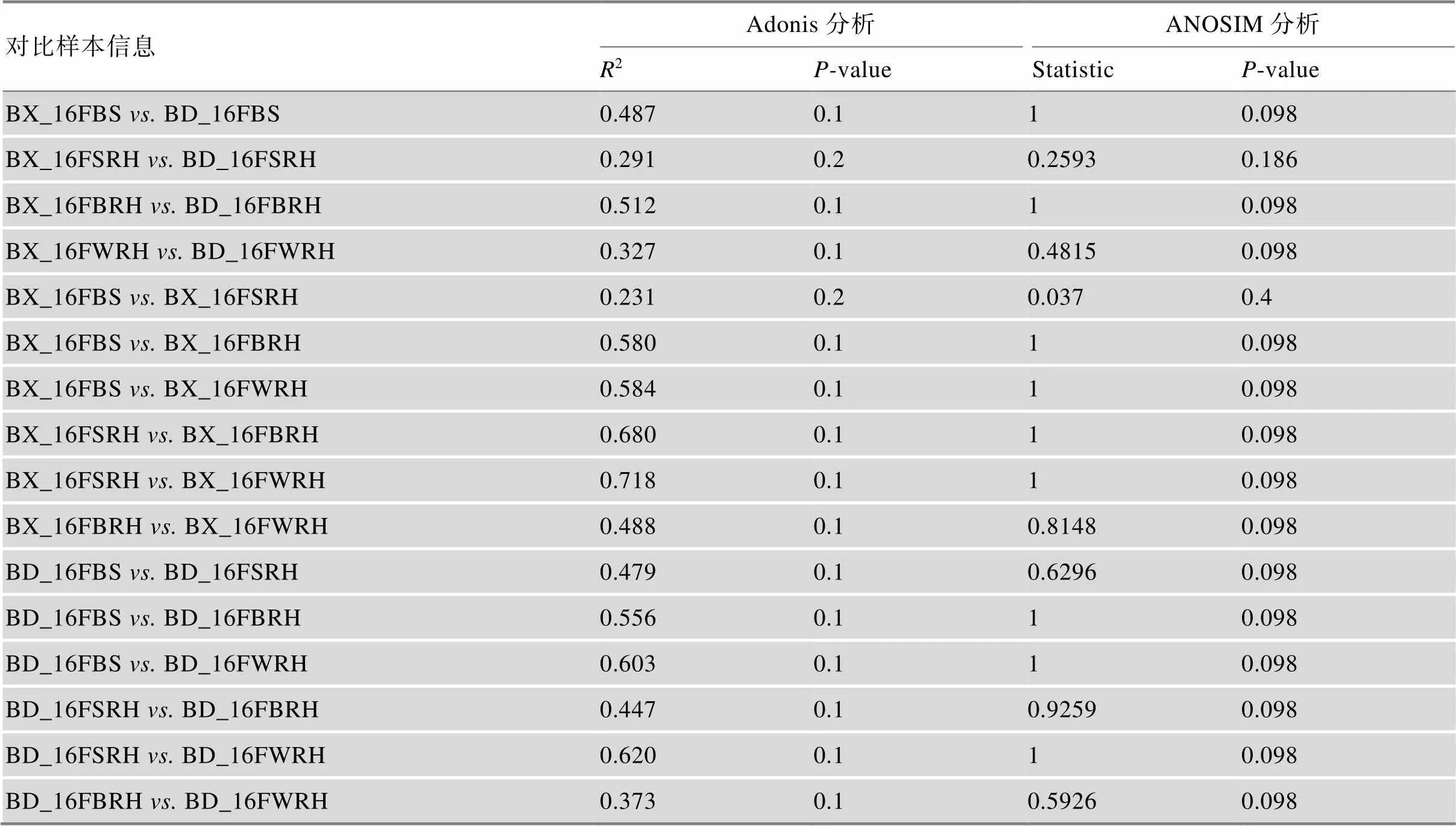
表1 不同样品细菌群落结构的统计分析
ANOSIM和Adonis基于Bray Curtis距离进行比较。详细处理信息同图1。>0.05表明,同一基因型大豆不同取样部位和同一取样部位不同基因型大豆的样品两两对比没有显著差异。
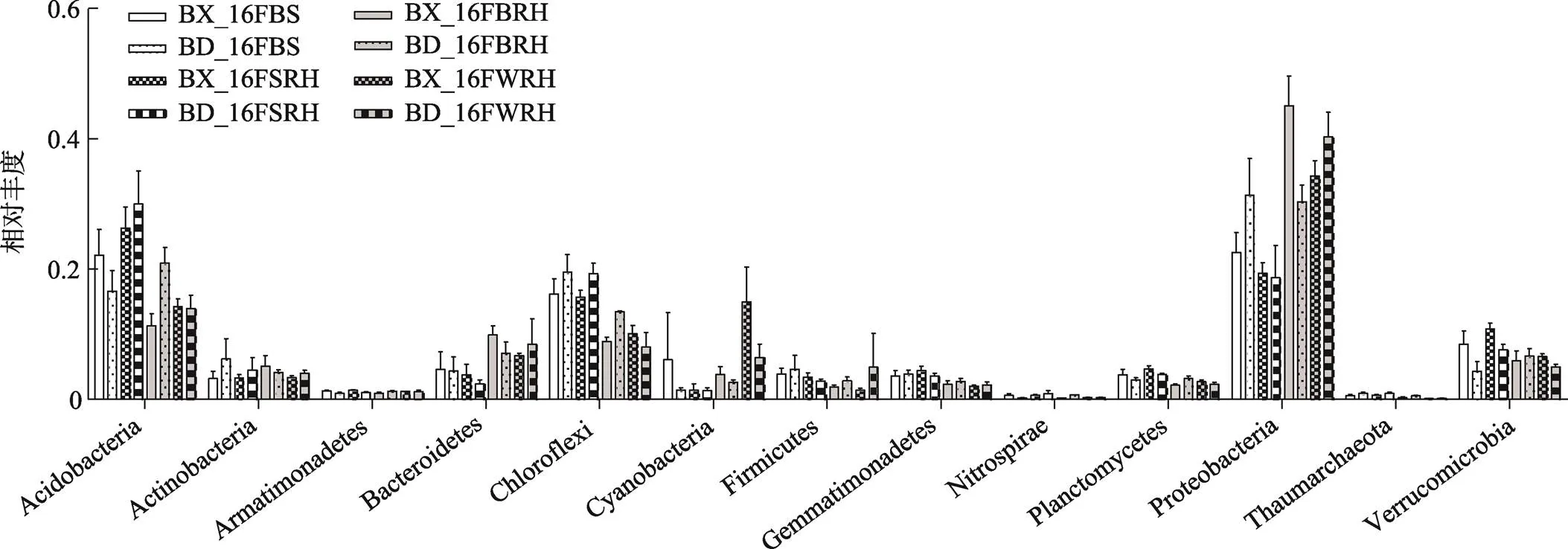
图3 各样本中相对丰富最高的13个主要门水平物种(>1%)
所选的13个主要门水平物种的相对丰度皆大于1%。详细处理信息同图1。
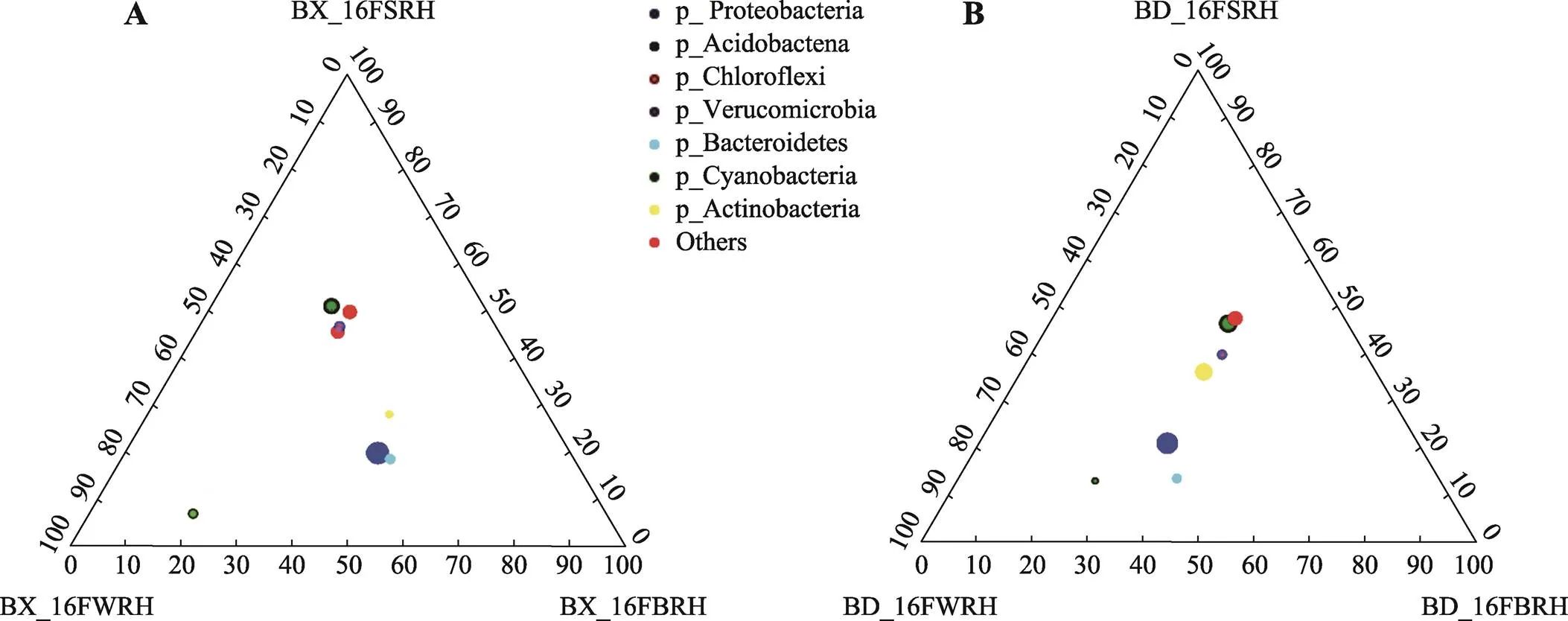
图4 门分类水平3个根际采样部位三元图
A:铝耐受型大豆BX10根际3个采样部位三元图;B:铝敏感型大豆BD2根际3个采样部位三元图。详细处理信息同图1。
2.4 铝敏感型大豆BD2富集与固氮直接相关的功能基因
利用PICRUSt软件进行16S rDNA的COG (clusters of orthologous groups of proteins,同源蛋白簇)功能预测和分析。如附图5所示,COG功能的分类组成及其丰度在各处理间并无显著差异。此外存在几种丰度较高的功能基因,如氨基酸转运与代谢(amino acid transport and metabolism)、细胞壁/膜/包膜生物发生(cell wall/membrane/envelope biogenesis)、信号传导机制(signal transduction mechanism)等。此外,从COG功能的相对丰度进行比较,COG0347、COG1348、COG1433、COG2710、COG3870、COG4656、COG5420、COG5456和COG5554等9个同源蛋白簇均可能与固氮直接相关(表3)。单因素方差分析进行显著性检验表明,相比于BX10,BD2在BRH中富集COG1433和COG3870,而在WRH中富集COG5420和COG5554。
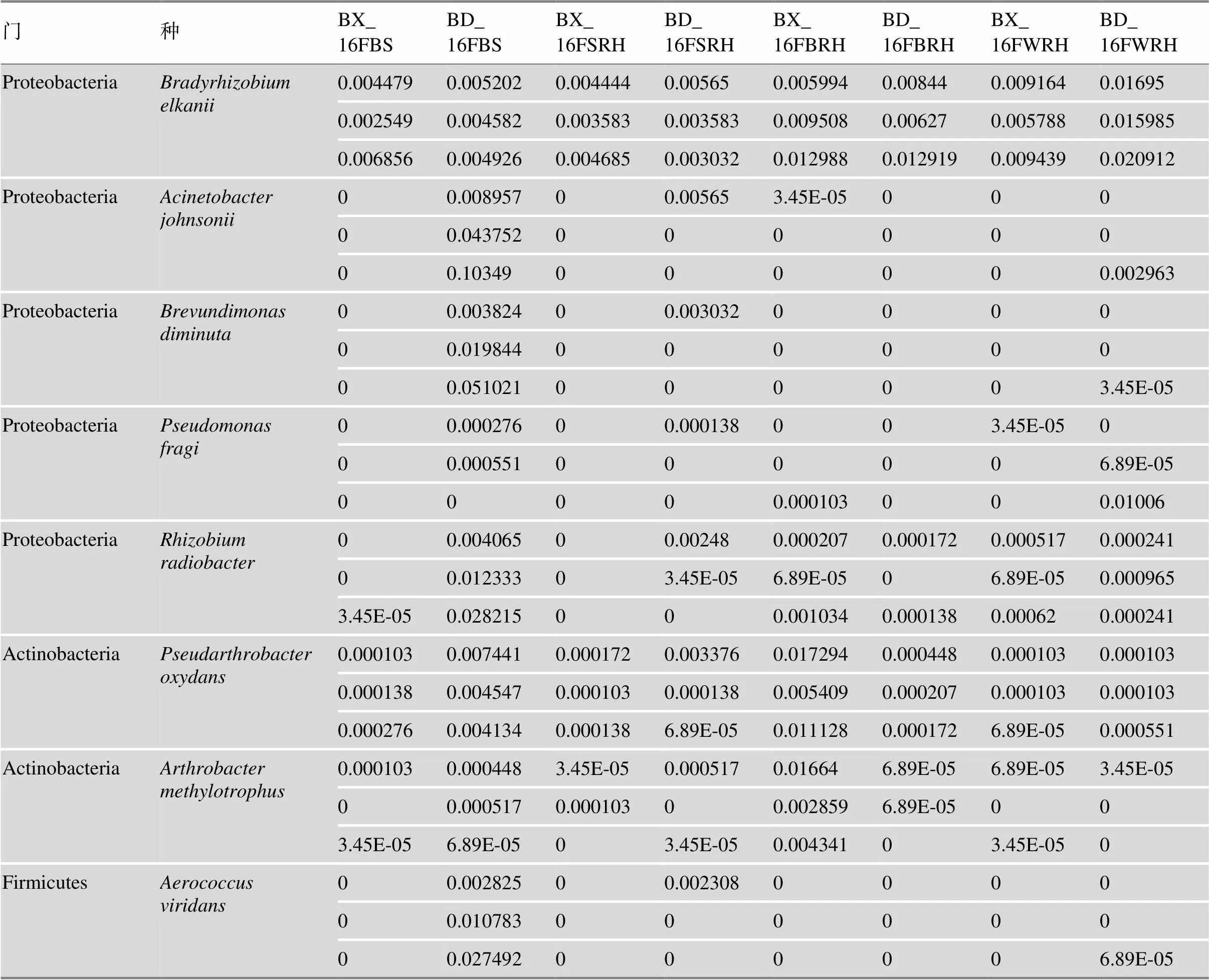
表2 种分类水平物种在各样本中的相对丰度
精确到种分类水平共找出8个种,属于变形菌门、放菌门和厚壁菌门。处理信息同图1。
3 讨论
本课题组之前的研究发现,耐铝大豆BX10和铝敏感大豆BD2诱导的根际土壤细菌和有机酸与群落功能有所不同[15,16]。具体体现在,根际土壤有机酸分析表明,BX10提高了柠檬酸的浓度,而BD2降低了柠檬酸的浓度;苹果酸仅在BX10根际土壤中存在。群落水平的生理分析表明,BX10对有机碳底物代谢能力的影响可能大于BD2。序列分析表明,两种大豆对一些根际细菌有刺激作用,如不动杆菌()、阿米巴假丝酵母()和未培养的蛋白杆菌(uncultured proteobacterium)。群落水平的生理分析表明,BX10对有机碳底物代谢能力的影响可能大于BD2。不仅如此,两种大豆不同时期分泌的有机酸不同,且在不同生长时期,真菌/细菌比例及革兰氏阴性/阳性菌的比例也各不相同。与BD2相比,BX10的磷脂脂肪酸在苗期和开花期具有较高的多样性,在结荚期有较低的多样性[15,16]。以上研究揭示了两种大豆根际细菌可能与其分泌有机酸有关。而本研究的目的,主要是通过分析高通量测序的结果,研究大豆基因型和宿主生态位(不同采样部位)对酸性红壤中大豆根际细菌群落组成和结构的影响。
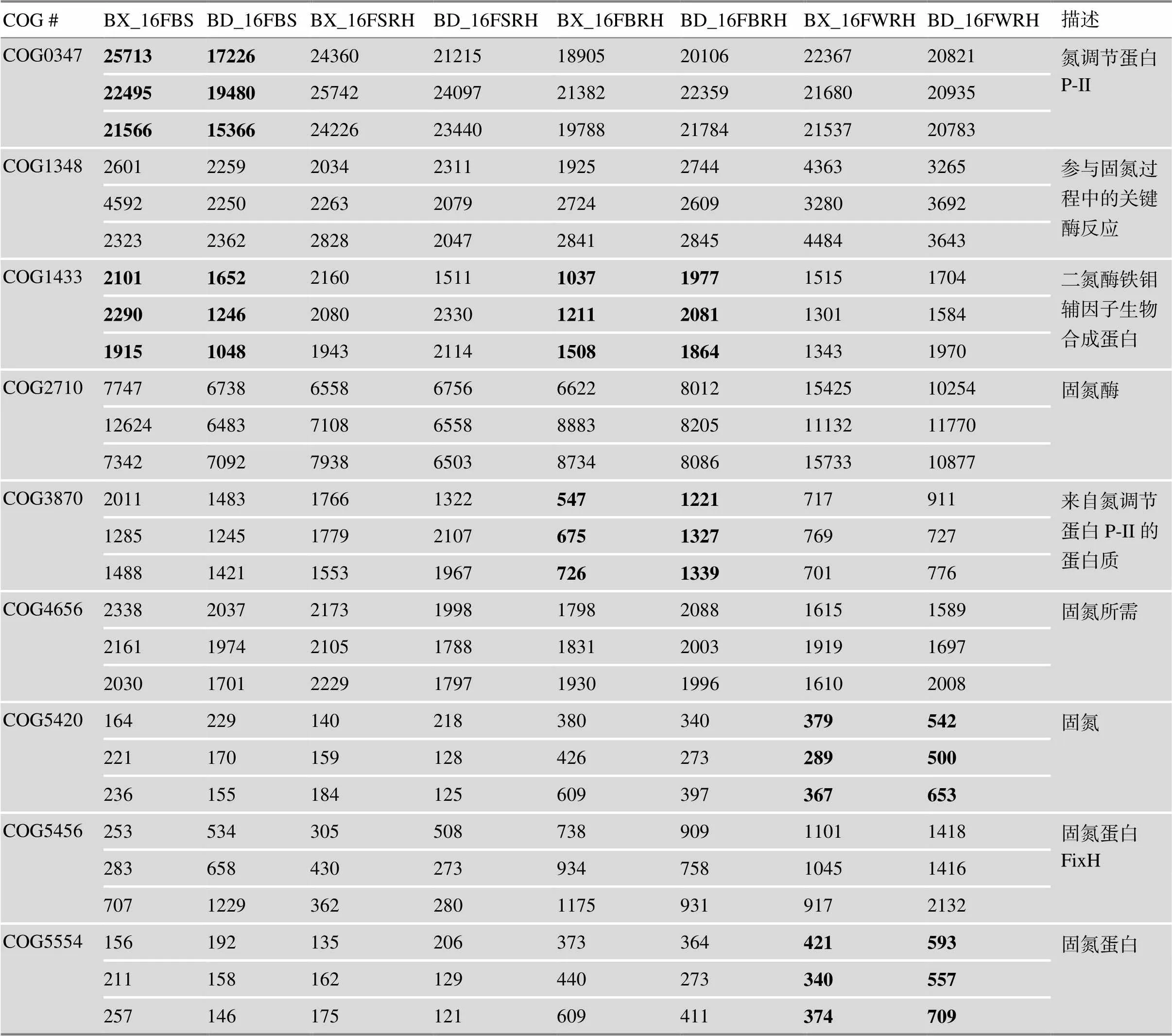
表3 固氮相关COG功能分类统计
处理信息同图1。显著性差异检验采用单因素方差分析,数字加粗表示BX10和BD2组之间存在显著差异(<0.05)。
本研究首先进行alpha多样性指数的分析,发现测序数据量合理且群落覆盖度较好。进行显著性差异分析发现除却BX_16FWRH在simpson指数上略高于BD_16FWRH外,剩下的各指数中BX10和BD2在相同采样部位的alpha多样性无显著性差异。甚至相同大豆(BX10或BD)对比不同采样部位也无显著性差异。对beta多样性分析的结果同样发现,发现在同一采样部位,BX10和BD2根际细菌群落的beta多样性在统计学意义上仍无显著性差异;同一基因型的4个不同取样部位的样品也无显著性差异。
Beta多样性是Whittaker于1960年提出的,定义为群落组成变化的程度,或群落分化的程度,与环境的复杂梯度或环境的模式有关[31]。其不仅可以反映样本之间的多样性距离关系,而且还可以反映生物群落之间的分化程度。影响细菌群落的beta多样性的因素很多,而细微的差别(各样本在具体某些属、种等的差异)往往会被掩埋在总体的无差异中。因此本研究中着重对比了各分类水平物种的组成和相对丰度。
本研究发现蓝细菌门在不同采样部位差异明显,特别是WRH部位极高。做三个根际采样部位三元图发现,两种基因型大豆的根部微生物群落,都存在蓝细菌门在WRH部位富集的情况,而这些细菌多为PGPR。目前已测序的能够固氮的微生物主要有:变形菌门、蓝细菌门、放线菌门、厚壁菌门、绿弯菌门、绿菌门(Chlorobi)及广古菌门(Euryarchaeota)[32~35]。蓝细菌门可以作为一种天然的生物肥料,在提高土壤肥力,进而提高植物性能方面发挥着重要作用。有文献证实,蓝细菌门可以帮助植物在胁迫环境下获得更好的形态、生理生化和抗氧化防御系统属性(生长特性、产量构成、光合效率,增强一系列酶活等)[36]。也就是说,BX10的根部对于蓝细菌,或者说这类可以增强植物抗逆性的PGPR,存在一定的富集作用,而BD2从三元图上来说也有富集的趋势,却在统计学意义上没有显著差异,这可以从侧面说明BX10,或者说可能是BX10的根系分泌物,增强了植物根部对某些有益菌的富集招募作用。也可能是因为所取样品重复数不足,误差的存在导致了显著性差异的不显著。
植物根际微生物群落主要由四种细菌组成:放线菌门、拟杆菌门、厚壁菌门和变形菌门[37~42]。本研究首先对厚壁菌门在不同样本中的丰度进行比较,结果显示不同样本中厚壁菌门丰度无显著变化,说明厚壁菌门在酸性土壤中丰度较低是一个普遍现象,而不是因为种植的大豆和大豆根系的环境引起的。芽孢杆菌目(Bacillales)属于厚壁菌门,且其中很多属种属于PGPR,结果表明各样本之间芽孢杆菌目的丰度无显著变化。最后对属分类水平一些主要的丰度有差异的物种进行分析(one-way ANOVA)。其他3种被广泛报道作为根际微生物群落的主要组成的放线菌门,拟杆菌门和变形菌门也进行了显著性差异分析,发现放线菌门在各样本间无差异,而拟杆菌门的BX_16FWRH和BX_16FBRH的丰度显著高于BX_16FSRH,变形菌门更是存在BX_16FWRH> BX_16FBRH>BX_16FSRH>BX_16FBS的现象,证明变形菌门受根部影响显著,呈梯度积累富集。
利用本课题组之前从酸性红壤中分离出一株根瘤菌(命名为)的16S rDNA 测序数据,在NCBI网站上进行比对发现和、以及聚为一支。因此本研究也研究了各采样部位根瘤菌属的丰度差异,根瘤菌属属于变形菌门(Proteobacteria),α变形菌纲(Alpha-proteobacteria)。我们发现同一采样部位两个基因型相比,根瘤菌属的丰度无显著性差异。这也从侧面说明根瘤菌在红壤中含量稳定,所以容易被分离鉴定出来。伯克氏菌属()属于变形菌门,β变形菌纲(Beta-proteobacteria),在BX_16FBRH中丰度高于所有其他样品。伯克氏菌属被报道与固氮相关,同时也有研究发现他们之中有的种属于耐铝细菌[43,44]。这可能和BX10相比BD2在酸性土壤中拥有较好的生长优势有关。此外,还存在嗜酸栖热菌属(属于放线菌门)在BD_16FBS显著高于其他样品的现象。以及粘液细菌属(属于拟杆菌门),存在FBRH在两个基因型都显著高于对应的FSRH (即BX_16FBRH>BX_16FSRH, BD_16FBRH>BD_16FSRH)。即在FBSH这个部位对属存在富集作用。
精确到种分类水平后我们找到8个种,分别进行了统计学分析。我们发现,的丰度在各样品之间无显著性差异,此菌与腐生代谢/食品腐败相关[45,46]。而在BD_16FWRH中的丰度远高于BD2的其他采样部位。,,和都发现在BD_16FBS的丰度远高于其他样本。其中属于共生固氮菌,它存在于植物的根瘤,甚至可以进入大豆的根瘤内部,发挥共生固氮作用。生物固氮提供了生物圈65%的可用氮,是世界上固定大气氮的最大来源[47];,参与反硝化作用(denitrification)[48,49];即可以作为人类的病原菌,也可以帮助植物缓解砷毒害及促进生长[50,51];是一种在环境中广泛传播的细菌,临床上这种细菌与动物和人类的不同疾病有关[52]。和的丰度则有BX_16FBRH大于BX10其他样品的情况。被报道与具有ACC脱氨酶活性的固氮菌序列相似性达到99.132%[53],则存在二甲基砜和二甲基亚砜还原酶活性,二甲基砜和二甲基亚砜在自然界中主要存在于海洋和土壤中,在植物生长中作为营养物质被吸收[54]。酸杆菌门虽然也是根际微生物群落的重要组成部分,但是精确到分类水平并未找到属于酸杆菌门的细菌[55]。三代16S rDNA全长测序相比于二代16S rDNA可变区片段的测序,其更长的读长的优势无疑也会在今后研究中起到更大的作用。
16S rDNA的功能预测与分析的结果表明,各样本在16S功能预测上组成和丰度一致。通过单因素方差分析比较了与固氮有直接关系的COG功能基因在各样本的丰度,研究发现在根外对照土中,BX10相比BD2富集COG0347和COG1348。这个现象可能是由于实验误差引起的,也有可能是因为耐铝大豆防止功能冗余而产生的对固氮菌的排斥作用,值得进一步分析。同时本研究还发现,BD2相比BX10在刷后根际土中富集COG1433和COG3870,在冲洗后的根际土中富集COG5420和COG5554。这可能是因为发现不同采样部位不同基因型各富集不同的与固氮有直接关系的COG功能基因铝敏感型大豆为了在酸铝环境中获得更好生长状态的一种补偿机制。
综上所述,各样本在alpha多样性,beta多样性上无显著性差异。在同一个基因型大豆不同采样部位间比较分析表明存在不同部位选择性富集不同的PGPR的现象。PCA和PCoA分析说明植物生长主要影响根际的BRH及WRH部位的微生物,对SRH部位影响较小,而BRH及WRH部位距离植物根的物理距离相对更近。门分类水平物种丰度进行比较表明,WRH部位存在富集蓝细菌门的现象。统计分析发现BX10根部对于增强植物抗逆性的PGPR有富集作用,包括蓝细菌门、拟杆菌门和变形菌门等,其中部分属种还与固氮和耐铝的功能相关,这也能部分解释为什么BX10可以在酸性红壤中获得更好的生长状态。最后,16S rDNA的COG功能预测和分析的结果表明,铝敏感型大豆BD2对于与固氮相关的功能基因存在富集作用,这可能与BD2为了在酸铝环境中生长的补偿机制相关,还有待进一步研究证实。
附加材料详见文章电子版www.chinagene.cn。
[1] Cheng FX, Cao GQ, Wang XR, Zhao J, Yan XL, Liao H. Isolation and application of effective nitrogen fixation rhizobial strains on low-phosphorus acid soils in South China., 2009, 54(3): 412–420.
[2] Kochian LV. Cellular mechanisms of aluminum toxicity and resistance in plants., 1995, 46, 237–260.
[3] Kochian LV, Hoekenga OA, Pineros MA. How do crop plants tolerate acid soils? Mechanisms of aluminum tolerance and phosphorous efficiency., 2004, 55: 459–493.
[4] Li SX. The current state and prospect of plant nutrition and fertilizer science., 1999, 5(3): 193–205.李生秀. 植物营养与肥料学科的现状与展望. 植物营养与肥料学报, 1999, 5(3): 193–205.
[5] Xu QP. Nitrogen cycle and nitrogen fixation., 2005, 21(3): 11–13.徐清平. 氮循环与固氮. 中学生物学, 2005, 21(3): 11–13.
[6] Foy CD. Plant adaptation to acid, aluminum-toxic soils., 1988, 19(7–12): 959–987.
[7] Zhang X, Liu P, Yang Y, Xu G. Effect of Al in soil on photosynthesis and related morphological and physiological characteristics of two soybean genotypes., 2007, 48(4): 435–444.
[8] Delhaize E, Craig S, Beaton CD, Bennet RJ, Jagadish VC, Randall PJ. Aluminum tolerance in wheat (L.) (I. Uptake and distribution of aluminum in root apices)., 1993, 103(3): 685–693.
[9] Vitorello VA, Capaldi FR, Stefanuto VA. Recent advances in aluminum toxicity and resistance in higher plants., 2005, 17: 129–143.
[10] Ryan PR, Delhaize E, Jones DL. Function and mechanism of organic anion exudation from plant roots., 2001, 52: 527–560.
[11] Yang ZM, Sivaguru M, Horst WJ, Matsumoto H. Aluminium tolerance is achieved by exudation of citric acid from roots of soybean ()., 2000, 110(1): 72–77.
[12] Avis TJ, Gravel V, Antoun H, Tweddell RJ. Multifaceted beneficial effects of rhizosphere microorganisms on plant health and productivity., 2008, 40(7): 1733–1740.
[13] Dong DF, Peng XX, Yan XL. Organic acid exudation induced by phosphorus deficiency and/or aluminium toxicity in two contrasting soybean genotypes., 2004, 122(2): 190–199.
[14] Zhen Y, Miao L, Su J, Liu SH, Yin YL, Wang SS, Pang YJ, Shen HG, Tian DC, Qi JL, Yang YH. Differential responses of anti-oxidative enzymes to aluminum stress in tolerant and sensitive soybean genotypes., 2009, 32(8): 1255–1270.
[15] Yang T, Ding Y, Zhu Y, Li Y, Wang X, Yang R, Lu G, Qi J, Yang Y. Rhizosphere bacteria induced by aluminum- tolerant and aluminum-sensitive soybeans in acid soil., 2012, 58(6): 262–267.
[16] Yang TY, Liu GL, Li YC, Zhu SM, Zou AL, Qi JL, Yang YH. Rhizosphere microbial communities and organic acids secreted by aluminum-tolerant and aluminum- sensitive soybean in acid soil., 2012, 48(1): 97–108.
[17] Li YC, Yang TY, Zhang PP, Zou AL, Peng X, Wang LL, Yang RW, Qi JL, Yang YH. Differential responses of the diazotrophic community to aluminum-tolerant and aluminum-sensitive soybean genotypes in acidic soil., 2012, 53: 76–85.
[18] Li YL, Fan XR, Shen QR. The relationship between rhizosphere nitrification and nitrogen-use efficiency in rice plants., 2008, 31(1): 73–85.
[19] Inceoğlu O, Salles JF, van Overbeek L, van Elsas JD. Effects of plant genotype and growth stage on the betaproteobacterial communities associated with different potato cultivars in two fields., 2010, 76(11): 3675–3684.
[20] Lu GH, Tang CY, Hua XM, Cheng J, Wang GH, Zhu YL, Zhang LY, Shou HX, Qi JL, Yang YH. Effects of an-transgenic soybean line ZUTS31 on root-associated bacterial communities during field growth., 2018, 13(2): e0192008.
[21] Lu GH, Zhu YL, Kong LR, Cheng J, Tang CY, Hua XM, Meng FF, Pang YJ, Yang RW, Qi JL, Yang YH. Impact of a glyphosate-tolerant soybean line on the rhizobacteria, revealed by Illumina MiSeq., 2017, 27(3): 561–572.
[22] Wen ZL, Yang MK, Du MH, Zhong ZZ, Lu YT, Wang GH, Hua XM, Fazal A, Mu CH, Yan SF, Zhen Y, Yang RW, Qi JL, Hong Z, Lu GH, Yang YH. Enrichments/derichments of root-associated bacteria related to plant growth and nutrition caused by the growth of an-transgenic maize line in the field., 2019, 10: 1335.
[23] Kennedy K, Hall MW, Lynch MDJ, Moreno-Hagelsieb G, Neufeld JD. Evaluating bias of illumina-based bacterial 16S rRNA gene profiles., 2014, 80(18): 5717–5722.
[24] Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, Gormley N, Gilbert JA, Smith G, Knight R. Ultra- high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms., 2012, 6(8): 1621–1624.
[25] Kozich JJ, Westcott S L, Baxter NT, Highlander SK, Schloss PD. Development of a dual-Index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform., 2013, 79(17): 5112–5120.
[26] Lu GH, Hua XM, Liang L, Wen ZL, Du MH, Meng FF, Pang YJ, Qi JL, Tang CY, Yang YH. Identification of major rhizobacterial taxa affected by a glyphosate-tolerant soybean line via shotgun metagenomic approach., 2018, 9(4): 214.
[27] Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky J R, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. QIIME allows analysis of high-throughput community sequencing data., 2010, 7(5): 335–336.
[28] Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. Introducing mothur: open-source, platform- independent, community-supported software for describing and comparing microbial communities., 2009, 75(23): 7537–7541.
[29] Bulgarelli D, Garrido-Oter R, Münch PC, Weiman A, Dröge J, Pan Y, McHardy AC, Schulze-Lefert P. Structure and function of the bacterial root microbiota in wild and domesticated barley., 2015, 17(3): 392–403.
[30] Liu YX, Qin Y, Guo XX, Bai Y. Methods and applications for microbiome data analysis., 2019, 41(9): 845–862.刘永鑫, 秦媛, 郭晓璇, 白洋. 微生物组数据分析方法与应用. 遗传, 2019, 41(9): 845–862.
[31] Whittaker RH. Vegetation of the siskiyou mountains, oregon and california., 1960, 30(4): 280– 338.
[32] Dos Santos PC, Fang Z, Mason SW, Setubal JC, Dixon R. Distribution of nitrogen fixation and nitrogenase-like sequences amongst microbial genomes., 2012, 13: 162.
[33] Franche C, Lindström K, Elmerich C. Nitrogen-fixing bacteria associated with leguminous and non-leguminous plants., 2008, 321(1–2): 35–59.
[34] Masson-Boivin C, Giraud E, Perret X, Batut J. Establishing nitrogen-fixing symbiosis with legumes: how many rhizobium recipes?, 2009, 17(10): 458–466.
[35] Peix A, Ramírez-Bahena MH, Velázquez E, Bedmar EJ. Bacterial associations with legumes., 2014, 34(1–3): 17–42.
[36] Zaki SS, Belal EEE, Rady MM. Cyanobacteria and glutathione applications improve productivity, nutrient contents, and antioxidant systems of salt-stressed soybean plant., 2019, 76: 72–85.
[37] Fierer N, Strickland MS, Liptzin D, Bradford MA, Cleveland CC. Global patterns in belowground communities., 2009, 12(11): 1238–1249.
[38] Bulgarelli D, Rott M, Schlaeppi K, van Themaat EVL, Ahmadinejad N, Assenza F, Rauf P, Huettel B, Reinhardt R, Schmelzer E, Peplies J, Gloeckner FO, Amann R, Eickhorst T, Schulze-Lefert P. Revealing structure and assembly cues forroot-inhabiting bacterial microbiota., 2012, 488(7409): 91–95.
[39] Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, Del Rio TG, Edgar RC, Eickhorst T, Ley RE, Hugenholtz P, Tringe SG, Dangl JL. Defining the coreroot microbiome., 2012, 488(7409): 86–90.
[40] Schlaeppi K, Dombrowski N, Oter RG, van Themaat EVL, Schulze-Lefert P. Quantitative divergence of the bacterial root microbiota ina relatives., 2014, 111(2): 585–592.
[41] Edwards J, Johnson C, Santos-Medellin C, Lurie E, Podishetty NK, Bhatnagar S, Eisen JA, Sundaresan V. Structure, variation, and assembly of the root-associated microbiomes of rice., 2015, 112(8): E911–920.
[42] Hu YL, Dai R, Liu YX, Zhang JY, Hu B, Chu CC, Yuan HB, Bai Y. Analysis of rice root bacterial microbiota of nipponbare and ir24., 2020, 42(5): 506–518.胡雅丽, 戴睿, 刘永鑫, 张婧赢, 胡斌, 储成才, 袁怀波, 白洋. 水稻典型品种日本晴和IR24根系微生物组的解析. 遗传, 2020, 42(5): 506–518.
[43] Minerdi D, Fani R, Gallo R, Boarino A, Bonfante P. Nitrogen fixation genes in an endosymbioticstrain., 2001, 67(2): 725–732.
[44] Huang SC, Wang XD, Liu X, He GH, Wu JC. Isolation, identification, and characterization of an aluminum- tolerant bacteriumsp. SB1 from an acidic red soil., 2018, 28(6): 905–912.
[45] De Filippis F, La Storia A, Villani F, Ercolini D. Strain-level diversity analysis ofafter in situ pangenome reconstruction shows distinctive spoilage-associated metabolic traits clearly selected by different storage conditions., 2018, 85(1): e02212–e02218
[46] Stanborough T, Fegan N, Powell SM, Tamplin M, Chandry PS. Vibrioferrin production by the food spoilage bacterium., 2018, 365(6).
[47] Lodwig EM, Hosie AHF, Bourdès A, Findlay K, Allaway D, Karunakaran R, Downie JA, Poole PS. Amino-acid cycling drives nitrogen fixation in the legume-Rhizobium symbiosis., 2003, 422(6933): 722–726.
[48] Zhang Y, Wang XJ, Wang WQ, Sun ZT, Li J. Investigation of growth kinetics and partial denitrification performance in strainunder different environmental conditions., 2019, 6(12): 191275.
[49] Wen G, Wang T, Li K, Wang HY, Wang JY, Huang TL. Aerobic denitrification performance of strainWGX-9 using different natural organic matter as carbon source: Effect of molecular weight., 2019, 164: 114956.
[50] Menuet M, Bittar F, Stremler N, Dubus JC, Sarles J, Raoult D, Rolain JM. First isolation of two colistin- resistant emerging pathogens,and, in a woman with cystic fibrosis: a case report., 2008, 2: 373.
[51] Singh N, Marwa N, Mishra SK, Mishra J, Verma PC, Rathaur S, Singh N. Brevundimonas diminuta mediated alleviation of arsenic toxicity and plant growth promotion inL., 2016, 125: 25–34.
[52] Liu G, Liu YX, Ali T, Ferreri M, Gao J, Chen W, Yin JH, Su JL, Fanning S, Han B. Molecular and phenotypic characterization ofassociated with subclinical bovine mastitis., 2015, 10(4): e0125001.
[53] Busse HJ. Review of the taxonomy of the genus, emendation of the genus, proposal to reclassify selected species of the genusin the novel generagen. nov.,gen. nov.,gen. nov.,gen. nov. andgen. nov., and emended description of., 2016, 66(1): 9–37.
[54] Borodina E, Kelly DP, Schumann P, Rainey FA, Ward-Rainey NL, Wood AP. Enzymes of dimethylsulfone metabolism and the phylogenetic characterization of the facultative methylotrophssp. nov.,sp. nov., andsp. nov., 2002, 177(2): 173–183.
[55] Li ZF, Feng ZL, Zhao LH, Shi YQ, Feng HJ, Zhu HQ. Effects of transgenic cotton expressing chitinase and glucanase genes on the diversity of soil bacterial community., 2015, 37(8): 821–827.李志芳, 冯自力, 赵丽红, 师勇强, 冯鸿杰, 朱荷琴. 转几丁质酶和葡聚糖酶双价基因棉花对土壤细菌种群多样性的影响. 遗传, 2015, 37(8): 821–827.
Bacterial composition, function and the enrichment of plant growth promoting rhizobacteria (PGPR) in differential rhizosphere compartments of Al-tolerant soybean in acidic soil
Zhongling Wen, Minkai Yang, Xingyu Chen, Chenyu Hao, Ran Ren, Shujuan Chu, Hongwei Han, Hongyan Lin, Guihua Lu, Jinliang Qi, Yonghua Yang
,,,,,
Low pH with aluminum (Al) toxicity are the main limiting factors affecting crop production in acidic soil. Selection of legume crops with acid tolerance and nitrogen-fixation ability should be one of the effective measures to improve soil quality and promote agricultural production. The role of the rhizosphere microorganisms in this process has raised concerns among the research community. In this study, BX10 (Al-tolerant soybean) and BD2 (Al-sensitive soybean) were selected as plant materials. Acidic soil was used as growth medium. The soil layers from the outside to the inside of the root are bulk soil (BS), rhizosphere soil at two sides (SRH), rhizosphere soil after brushing (BRH) and rhizosphere soil after washing (WRH), respectively. High-throughput sequencing of 16S rDNA amplicons of the V4 region using the Illumina MiSeq platform was performed to compare the differences of structure, function and molecular genetic diversity of rhizosphere bacterial community of different genotypes of soybean.The results showed that there was no significant difference in alpha diversity and beta diversity in rhizosphere bacterial community among the treatments. PCA and PCoA analysis showed that BRH and WRH had similar species composition, while BS and SRH also had similar species composition, which indicated that plant mainly affected the rhizosphere bacterial community on sampling compartments BRH and WRH. The composition and abundance of rhizosphere bacterial community among the treatments were then compared at different taxonomic levels. The ternary diagram of phylum level showed that Cyanobacteria were enriched in WRH. Statistical analysis showed that the roots of Al-tolerant soybean BX10 had an enrichment effect on plant growth promoting rhizobacteria (PGPR), which included Cyanobacteria, Bacteroides, Proteobacteria and some genera and species related to the function of nitrogen fixation and aluminum tolerance. The rhizosphere bacterial community from different sampling compartments of the same genotype soybean also were selectively enriched in different PGPR. In addition, the functional prediction analysis showed that there was no significant difference in the classification and abundance of COG (clusters of orthologous groups of proteins) function among different treatments. Several COGs might be directly related to nitrogen fixation, including COG0347, COG1348, COG1433, COG2710, COG3870, COG4656, COG5420, COG5456 and COG5554.Al-sensitive soybean BD2 was more likely to be enriched in these COGs than BX10 in BRH and WRH, and the possible reason remains to be further investigated in the future.
acidic soil; aluminum-tolerant soybean; rhizosphere; bacterial community; PGPR
2020-11-30;
2020-12-29
国家重点研发计划项目(编号:2016YFD0101005)、国家自然科学基金项目(编号:31870495, 31372140)、教育部创新团队项目(编号:IRT_14R27)资助[Supported by the National Key Research and Development Program of China (No. 2016YFD0101005), the National Natural Science Foundation of China (Nos. 31870495, 31372140), and the Program for Changjiang Scholars and Innovative Research Team in University from the Ministry of Education of China (No. IRT_14R27)]
文钟灵,博士研究生,研究方向:土壤分子微生物学。E-mail: DG1730028@smail.nju.edu.cn
杨旻恺,博士研究生,研究方向:土壤分子微生物学。E-mail: minkaiyang@163.com
文钟灵和杨旻恺并列第一作者。
杨永华,教授,博士生导师,研究方向:分子代谢与生物技术安全。E-mail: yangyh@nju.edu.cn
10.16288/j.yczz.20-409
2021/1/28 10:45:54
URI: https://kns.cnki.net/kcms/detail/11.1913.r.20210127.1604.002.html
(责任编委: 孔凡江)
