表皮松解型掌跖角化症的研究进展
2021-05-19林雪霏隋长霖
何 丽,陈 慧,林雪霏,李 曼,隋长霖,朱 威
掌跖角化症是一组以掌跖表皮角化过度为主要特征的常染色体显性遗传性皮肤病,该病可分为10多个型别,在全世界多个种族中均有发病。临床上将掌跖角化症分为非综合征型和综合征型,非综合征型中最常见的是表皮松解型掌跖角化症(epidermolytic palmoplantar keratoderma,EPPK),又 称 沃 纳 氏(Vorner)掌跖角化病,它由Vorner 于1901 年首次描述[1]。文献报道EPPK 患病率为每10 万人中1.0 ~4.4人[2,3],且发病无性别差异。该病以手掌和足跖弥漫性角化过度为主要临床表现,一些患者可能有多汗症、指节垫、手指弯曲和毁损[4]。女性患者患卵巢癌或乳腺癌的风险可能较正常人增加[5]。目前认为角蛋白基因突变是该病的致病基础。本文对EPPK 发病机制、临床表现及治疗措施进行综述。
1 发病机制
研究认为遗传因素、环境因素与EPPK 发病有关,其中分子遗传学因素为其主要病因。角蛋白为中间丝家族的重要成分,是表皮及毛发角质形成细胞的主要组成蛋白,也是多种细胞的骨架蛋白,可为上皮细胞提供结构支撑,也是细胞分化的重要标志。这些角蛋白分子有两种型别:1 型有28 种,2 型有26 种,已报道至少有54 种有功能的角蛋白分子,其中有18种角蛋白基因突变可引起各种各样的疾病。EPPK 的主要致病基因之一是角蛋白家族的角蛋白9(keratin9,KRT9)基因,KRT1、KRT10 和KRT16 等基因突变也可引起EPPK[6,7]。KRT9 基因序列全长4492bp,含8 个外显子,cDNA 全长2290 bp。KRT9 基因编码含623 个氨基酸的KRT9,KRT9 由螺旋杆状结构域、非螺旋的头部结构域及非螺旋的尾部结构域三部分构成[8]。螺旋杆状结构域包括4 个α 螺旋(1A、1B、2A 和2B)及螺旋之间的连接区域(L1、L12、L2)[9]。杆状结构域的1A 区为螺旋起始基序(HIM),2B 区为螺旋终末基序(HTM)[10]。
1.1 KRT9 基因突变
据文献报道,已在100 个EPPK 家族中鉴定出KRT9 基因的33 个突变(表1)[11],错 义 突 变 有30 个,其中23 个位于1A 区域(突变热点区域),7 个位于2B区域,绝大多数位于第1 外显子[12],其余3 个为同义突变[11]。综上,角蛋白基因突变集中在KRT9 基因编码的K9 螺旋杆状结构域的1A和2B 区 域,尤 其 是1A 区域,而该区域影响角蛋白网络结构的形成,可导致严重的临床表现[13]。据权威的国际人类中间丝数据库不完全统 计,在100 多 个EPPK 家系或散发病例中,KRT9 基因突变频率最高为位于第1外显区的p.Arg163Trp,约占33.4%。 此 外,p.Arg163Gln突变约占10.7%,p.Asn161Ser突变约占7.1%,p.Met157Val突变约占4.5%,前述4 种KRT9 突 变 型 占EPPK 基 因突变总数的55.7%[14]。另外,Li 等[8]报道了1 个三代中国家系的2 例EPPK 患者,对先证者及其儿子外显子测序,发现KRT9 基因中的杂合错义致病突变c.488G>A(p.Arg163Gln),该病例报告描述了导致中国EPPK的c.488G>A(p.Arg163Gln)突变,该突变的发现拓宽了EPPK 中KRT9 基因致病突变的范围。Chen 等[7]报道了1 个四代中国汉族人EPPK 家系,发现KRT9基因第1 外显子一个新的杂合错义突变488G>T。Li等[11]在3 个中国EPPK 家系中,检测到KRT9 基因位于编码保守中央α 螺旋杆状结构域中的3 个杂合突变(p.N161S,p.R163W和p.R163Q),这项研究证实了密码子163 是KRT9的热点突变位点,同时还发现p.N161S(4%)和p.R163W(4%)是与指节垫相关EPPK 的潜在热点突变,而与指节垫无关的EPPK 热点突变为p.R163Q。此外,KRT9 基因突变与患者掌跖角蛋白的表达亦有关系,Liu 等[15]报道1 个EPPK五代家系,在KRT9 基因螺旋起始序列中发现杂合突变c.T491C(p.L164P),且患者手掌KRT16/KRT6 蛋白表达显著增加,表明应激反应和伤口愈合的细胞角蛋白被活化。同时还发现患者KRT9/KRT2 表达增加,KRT10/KRT1 表达无变化,KRT14/KRT5 未检测到。作者首次报道了EPPK 患者中KRT9的p.L164P杂合突变与多种角蛋白表达的改变有关。
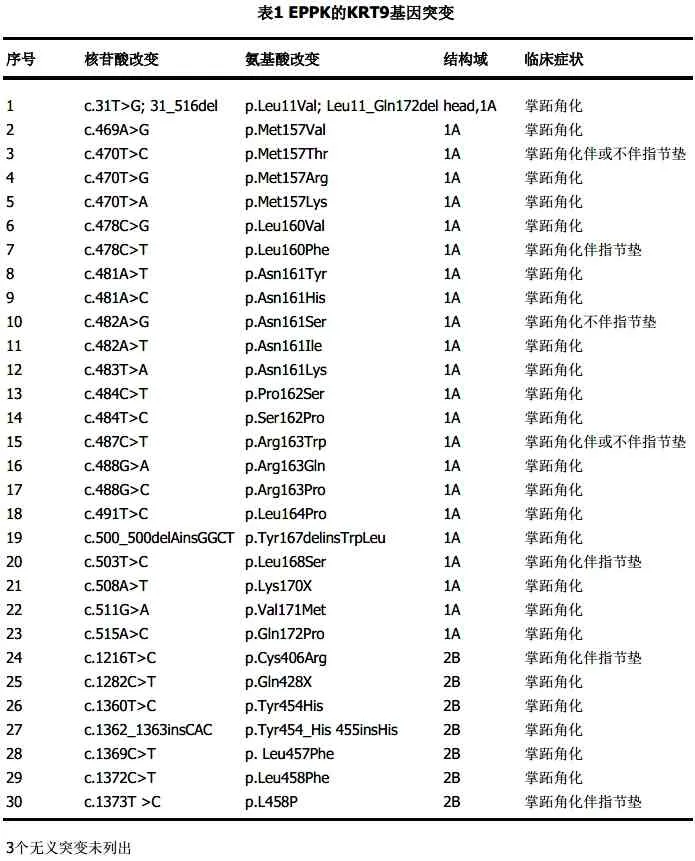
?
1.2 表型异质性
目前中国EPPK 家系患者中,除显示出典型的临床和组织病理学特征外,还有3 种主要的伴随症状:指节垫、与摩擦有关的皮肤损害、手掌挛缩。EPPK基因突变在不同人群中均有发生,但角化严重程度等临床症状与伴随症状总是不尽相同。尽管约60%的序列具有相同性,即使在携带相同突变的情况下,不同家系或不同个体之间仍存在表型差异。同一角蛋白基因突变的严重程度不仅取决于角蛋白中蛋白结构域的位置,而且同一位置特定氨基酸被替代也对其有影响。这种多样性可能与环境或表观遗传因素的不同有关,这需要对有特定KRT9 基因突变的动物模型进行深入研究。有文献报道了22 例KRT9 基因突变并且有伴随症状的EPPK 患者,发现15 例(68%)患者出现指节垫,这是最常见的伴随症状。而在这些患者中有11 例KRT9 基因突变分布于1A ~2B 结构域,没有特定的突变位点[11]。在指节垫中发现KRT9的异位表达,这可能与指关节垫的形成有关[16]。在目前已知的33 个KRT9 基因突变中,有15 个与指节垫有关。摩擦相关的皮肤损害(18%)发生在有p.L160F和p.R163W突变的中国人、日本人和印度人中[17,18]。
2 临床表现及组织病理特征
2.1 临床表现
EPPK 患者在出生后数周到数月即可逐渐出现典型临床症状,并持续终生。轻度患者仅有掌跖的表皮粗糙,大部分患者主要表现为双侧掌跖部位弥漫性角质增厚,色黄,在增厚的表皮周边有明显的红斑。患者手掌和足跖易皲裂,可伴疼痛感,症状严重时手指活动困难。此外,患者手和足的背部、手指、胫骨前、肘部和膝盖也可出现皮损,并持续终生。多数患者伴多汗、臭汗症状,某些患者还可伴随指节垫、断指或手指畸形等。通常,除轻度瘙痒外,患者无其他主观症状。
2.2 组织病理特征
EPPK 发病与角蛋白、角质化包膜、内聚力、细胞间通讯和跨膜信号转导蛋白等有关。该病组织病理学特征是角化不全、表皮溶解性角化过度、角质形成细胞明显的核周空泡化及位于棘状上层的巨大角质透明颗粒[19]。在超微结构上,EPPK 表现为异常的核周张力丝团块,并存在较大且变形的角质透明蛋白颗粒。频繁用手、机械摩擦引起表皮过度生长,电镜下观察有角蛋白和张力丝的异常聚集物,这些均可使表皮的稳定性降低,而这种过度生长的病态表皮可能是皮肤稳定性降低的一种代偿。
3 治疗措施
除一般的对症治疗,如皮损局部化学封包、药物浸泡和全身治疗如口服类维生素等[20],EPPK 尚无有效的治疗方法。临床和实验室观察均证实,角蛋白对维持表皮细胞间粘附具有重要作用,当正常组织动态平衡受到干扰时,如在伤口愈合和癌症期间,角蛋白起着重要的非机械作用。翻译后修饰,如角蛋白的糖基化和磷酸化,均在保护上皮细胞免受损伤中起重要作用。此外,角蛋白在调节免疫应答中也起作用。角蛋白相关疾病目前研究重点是开发新的治疗方法,包括通过突变角蛋白和小分子对应的小干扰RNA(siRNA)调节角蛋白的表达。目前EPPK 治疗的研究聚焦于变异组学,靶向siRNA 敲除EPPK 患者突变基因是一种有潜力的治疗方法[20]。Lyu 等[21]构建了一种敲入式KRT9基因突变EPPK 小鼠模型,模仿人类EPPK 患者,作者为EPPK 小鼠开发了一种突变体特异性短发夹RNA(shRNA)。使用萤光素酶报告基因测定法在体外鉴定了突变体特异shRNA,并递送至KRT9+/mut小鼠中,shRNA 介导的突变蛋白敲低,可使小鼠爪子皮肤的形态和功能几乎正常,而相同的shRNA 对野生型K9 小鼠几乎无作用,这提示可以尝试通过基因疗法治疗EPPK,这对未来的临床应用具有重要意义。Wang 等[14]报道了一个维吾尔族的六代EPPK 家系,包括4 个近亲婚姻家庭,发现与非近亲结婚的患者相比,近亲结婚患病人数更多,表型更严重,发病年龄更早。KRT9 基因突变杂合外显率为100%,生育患病子代的遗传风险高达50%,避免EPPK 家系的近亲婚姻对降低患病率至关重要。总而言之,变异组学、生殖遗传学和优生学的研究对预防EPPK 患儿的出生具有重要意义。
4 小结与展望
EPPK 是一种少见的常染色体显性遗传病,对患者日常生活、工作、身心健康等均有严重影响。本病临床症状体征相对典型,但发病机制未完全明确,主要涉及分子遗传学方面。角蛋白基因突变是本病的研究热点,患者临床表型与基因突变、环境因素等的相关性也有待深入的研究。本病目前尚无根治方法,基因诊断和产前诊断对预防该病发生和降低出生缺陷有重要意义。目前国内外报道的EPPK 多为散发或小家系,缺乏大宗临床研究,提示临床医生需重视大样本量临床病例的收集,以期为EPPK 的发病机制、治疗和预防提供新的思路。
