钙钛矿太阳电池中各功能层的光辐照稳定性研究进展*
2021-05-14李燕贺红党威武陈雪莲孙璨郑嘉璐
李燕 贺红 党威武 陈雪莲 孙璨 郑嘉璐
1) (西安石油大学材料科学与工程学院, 西安 710065)
2) (陕西国防工业职业技术学院智能制造学院, 西安 710300)
钙钛矿太阳电池简单的制备工艺、低廉的成本和优异的性能使其有望替代已产业化的硅太阳电池, 革新现有能源供给结构, 然而, 钙钛矿太阳电池的稳定性差制约了其产业化进程, 本文分别介绍了光辐照下, 钙钛矿薄膜内部本征的离子迁移行为和由此产生的磁滞、荧光淬灭/增强和电池失效问题; 以及典型的TiO2/钙钛矿界面的紫外光不稳定、空穴传输层和金属电极不稳定问题.作为依光器件深刻理解其光辐照稳定性对顺利解决电池各种环境稳定性问题至关重要.
1 引 言
能源枯竭和环境污染使人们越来越重视可持续发展, 实现人与自然和谐共处成为各国政府热切关注的问题.发展新型清洁能源缓解当前环境和能源问题是可持续发展的重要内涵.太阳能作为所有能量的源头, 其取之不竭, 用之不尽的特点, 使光能到电能的转换和利用成为可靠且有力的能源供给方式.近几十年, 科学研究的结果表明光伏器件,比如硅太阳电池, 具有低成本、易制造且高转换效率的巨大潜力.
在硅太阳电池之后, 研究者从未停止对更廉价、更高效光伏转换材料的探索, 比如, CuInGaSe材料[1,2]、有机小分子材料[3,4]、杂化钙钛矿材料[5,6].其中, 杂化钙钛矿材料是一类具有ABX3钙钛矿相的材料, 2009 年首次被应用到光伏电池, 当时所制备的钙钛矿太阳电池(perovskite solar cells, PSCs)仅有3.8%的转换效率, 但经过10 年的发展, 到2020 年其光电转换效率已高达25.5%[7], 电池组件的最大面积可达808 cm2[8].PSCs 在光电性能上的突飞猛进, 可归因于杂化钙钛矿材料对可见光谱的强吸收、载流子迁移率高且长、缺陷容忍能力强以及优异的双极性电荷输运能力等[9,10].当前围绕PSCs 产业化所面临的问题包括: 1) PSCs 在空气环境中的长期稳定性; 2)制备工艺中的溶剂绿色化; 3)开发新型廉价空穴传输材料; 4)高通量规模化的量产技术等.
相比较而言, 电池的稳定性问题被认为是其商业化的突出障碍, 实验室报道的PSCs 的最长稳定输出为1 年[11], 这远远达不到电池室外应用所要求的25 年.最初的研究进展主要集中在钙钛矿材料的氧气和水分不稳定问题, 随着对PSCs 磁滞现象的深入研究, 人们逐渐意识到钙钛矿材料是电子离子混合导体, 光辐照下的离子迁移及其与缺陷态的相互作用, 使得钙钛矿薄膜出现诸如光修复、光解、光致相分离等现象.除此之外, 研究发现当光和外部因素(比如, 氧气和水分)耦合作用时, 将加速电池失效进程, 甚至光可作为失效的主导因素[12,13].
所以, 本文以钙钛矿薄膜为中心, 综述了光辐照下其内部本征的离子迁移行为, 以所产生的磁滞、荧光淬灭/增强和钙钛矿失效现象为顺序, 展示了离子迁移与缺陷态的相互作用后出现的光修复、光解和光致相分离等现象, 此外, 本文综述了PSCs 中代表性的TiO2/钙钛矿界面紫外不稳定问题, 空穴传输材料和金属电极的光不稳定问题, 以期为PSCs 的长期稳定性做出贡献.
2 钙钛矿太阳电池的结构、工作原理及钙钛矿薄膜的特性
2.1 基本结构
PSCs 主要分为正向和反向两类结构, 如图1(a)和图1(c)所示[14], 在正向结构电池中, 各功能层能级的差异使电子流向透明导电半导体汇流极(通常为FTO(F:SnO2)或者ITO(Sn:In2O3)), 空穴流向金属汇流极; 在反向电池结构中, 各功能层能级的差异使空穴流向透明导电半导体汇流极, 电子流向金属汇流极.除此之外, 早期PSCs 起源于全固态染料敏化太阳能电池, 其基本结构中常使用几百纳米厚的TiO2多孔层.2012 年, Etgar 等[15]发现了钙钛矿材料的双极性电荷传输能力, 基于此设计的无介孔电子传输层的PSCs 甚至获得了7%的转换效率, 因此, 出于降低商业化成本的意图, 无介孔电子传输层的面结PSCs 得到了广泛研究.
2.2 工作原理
无论正向还是反向电池, 其工作原理均由钙钛矿薄膜捕获光子产生激子(高能电子-空穴对), 在各个功能层能级高低的诱导下, 电子被注入到电子传输层, 空穴被注入到空穴传输层, 进一步, 分离后的电子和空穴分别通过两侧的收集电极进入外电路, 实现对外供电.短路时, PSCs 内部PN 结的能级差为零, 电池对外输出最大电流, 即短路电流(short circuit density,Jsc); 开路时, PSCs 内部无能级差, 电池对外表现为最大电压, 即开路电压(open circuit voltage,Voc), 见图1(d)和1(e)[14].
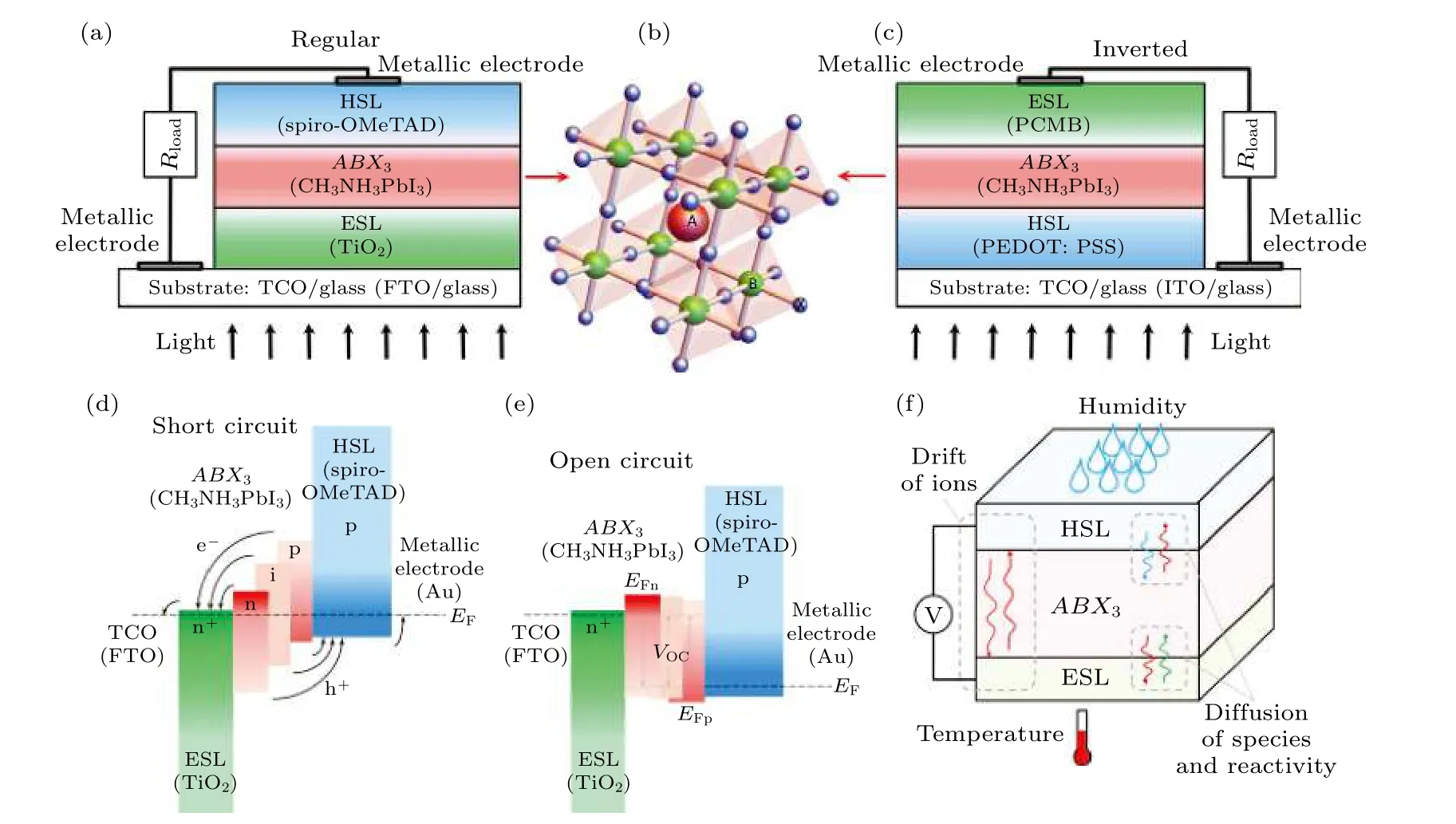
图1 PSCs 的(a)正向和(c)反向基本结构; (b)钙钛矿材料的晶体结构; 在(d)短路和(e)开路状态下PSCs 内部的能级结构;(f) PSCs 不稳定的主要诱因示意图[14]Fig.1.Basic structure of PSCs in (a) regular and (c) inverted configurations; (b) crystalline structure of perovskite materials; general energy band diagram at (d) short circuit and (e) open circuit; (f) main contributing factors in the degradation processes of PSCs[14].
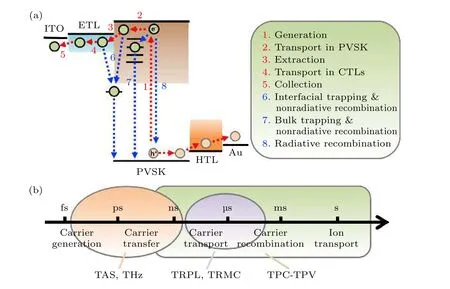
图2 PSCs 内部电荷-载流子动力学过程 (a)载流子动力学过程; (b)动力学过程发生的时间尺度[37]Fig.2.General charge-carrier processes within a PSC: (a) Carrier dynamics processes; (b) corresponding time scale of the cell[37].
PSCs 内部电荷到载流子的动力学过程包括正向过程: 1)电荷产生; 2)电荷在钙钛矿薄膜内传输;3)电子和空穴分别被电子传输层和空穴传输层提取; 4)电子和空穴在电子和空穴传输层内传输;5)电子和空穴被两侧的收集极收集.反向过程:6)界面复合; 7)体相复合和8)辐射复合.从时间维度上看, 电荷产生发生在飞秒量级, 电荷转移发生在皮秒量级, 电荷传输发生在纳秒量级, 电荷复合发生在微秒到毫秒量级, 离子迁移发生在秒量级.发生在不同时间维度的动力学过程可以用瞬态光吸收谱(transient absorption spectroscopy,TAS)[16,17]、太赫兹泵浦探针技术(terahertz pumpprobe technology, THz)[18−20], 时间分辨荧光光谱(time-resolved photoluminescence, TRPL)[21−25],时间分辨微波传导率(time-resolved microwave conductivity, TRMC)[26−28]、瞬态光电流(transient photocurrent, TPC)和瞬态光电压(transient photovoltage, TPV)测试[29−36], 如图2 所示[37].
2.3 钙钛矿薄膜的特性
PSCs 的核心部件是钙钛矿薄膜, 钙钛矿薄膜由ABX3单元组成, 其晶体结构如图3(a)和图3(b)所示[38].A位通常是有机阳离子, 如甲胺(MA+))或甲脒((FA+));B位是二价金属阳离子, 如Pb2+, Sn2+和Ge2+;X位是阴离子,如I–, Cl–或Br–.B位原子和6 个X位原子协调,形成一个八面体, 其中B在中心位置.8 个BX6八面体通过共享X点位相互连接, 构成一个立方晶格骨架, 骨架中心被原子A占据.因为B原子与X原子之间由极性共价键相互连接, 因此BX6八面体的结构比较稳定.事实上, 大量研究结果也表明了ABX3结构的稳定性取决于BX6框架和A原子之间的相互作用, 比如: 1)两个BX6八面体之间极易发生扭曲和旋转, 如图3(c)所示, 扭转使得钙钛矿材料的对称性发生了明显改变.2)占据骨架中心位置的A原子亦对钙钛矿晶体结构产生重要影响, 如果原子A太小, 钙钛矿结构坍塌, 如果原子A太大, 则角共享框架将被破坏, 如图3(d)—(f)所示.
对ABX3钙钛矿材料的长期研究形成了晶体结构预测的一定公共准则, 比如, 钙钛矿的结构及其稳定性可以由公差因子t和八面体因子µ决定.
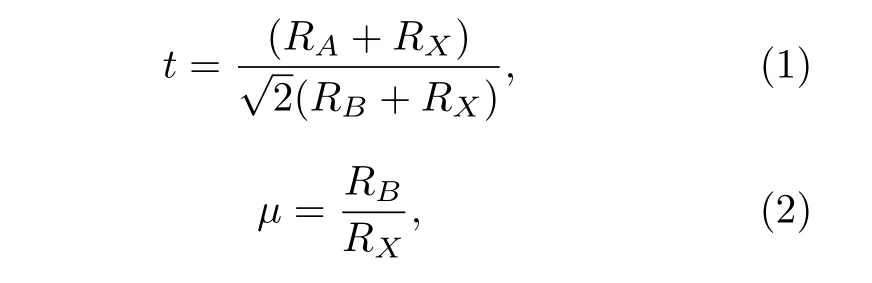
其中RA,RB和RX分别是原子A,B和X的半径.通常结构稳定的钙钛矿晶体t在(0.81—1.11)的区间,µ在(0.44—0.90)的区间.
钙钛矿材料具有较大的介电常数和较小的激子结合能[39,40], 这使光活化的激子在室温下即可分离, 在皮秒时间内可形成自由载流子, 且钙钛矿晶体结构中不存在类似PbI2空位和MAI 空位的Schottky 缺陷, 最可能存在的缺陷是Pb, I 和MA空位形成的位于带边位置的浅能级Frenkel 缺陷[20],这样的浅能级缺陷使钙钛矿材料表现出长电子空穴扩散长度, 以及良好的PN 双极性电荷传输能力, 因此钙钛矿材料的内量子效率可接近100%[41,42].
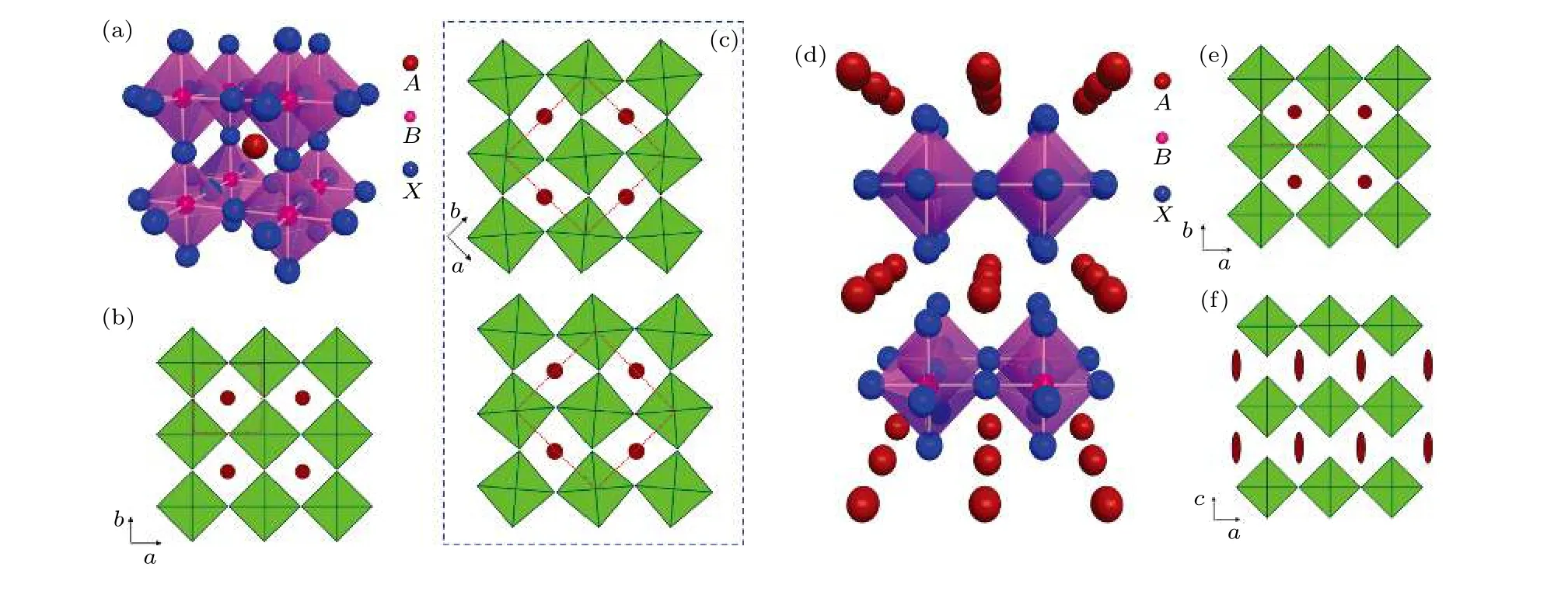
图3 ABX3 钙钛矿晶体结构的变化 (a), (b)标准钙钛矿结构; (c) BX6 八面体的扭曲和旋转引起的结构变化; (d)—(f)过大的A 位原子对钙钛矿结构的破坏[38]Fig.3.Structure of the ABX3: (a), (b) Standard perovskite structure; (c) structural changes caused by the twist and rotation of BX6 octahedron; (d)–(f) destruction of perovskite structure by too large A atom[38].
载流子作为转换过程中的能量载体, 其存活寿命、扩散长度、热力学分布等属性是决定PSCs 光电转换性能的重要因素[43−45].现在研究者已意识到PSCs 所使用的钙钛矿材料特殊的离子-电子混合导电机制[19], 在众多影响电池稳定性的因素中,光照引起的钙钛矿材料的离子迁移、TiO2/钙钛矿界面的分解失效、空穴传输材料的组分变化和金属电极的元素迁移是本征的, 并且不能通过电池封装消除掉, 因此深入认识这些现象及其对PSCs 性能的影响, 对PSCs 的高效化和稳定化都具有重要意义.
3 钙钛矿薄膜的光照不稳定的原因、现象、实验证据和抑制方法
3.1 钙钛矿薄膜的离子迁移
在MAPbI3中任何组成离子都可能迁移, 包括I–, Pb2+和MA+[46,47].活化能EA可用来衡量迁移的难易程度, 其决定于材料的晶体结构、离子半径、离子跳跃距离和离子携带电量等因素, 一般情况下, 晶格间隙位置大、离子电量小、跳跃距离短,越容易发生离子迁移.Mosconi 等[21]推算离子迁移的活化能在几百个meV 量级, Hoke 等[48]使用荧光光谱测试计算得到块体MAPbI3–xBrx的I–迁移EA为(0.27 ± 0.06) eV.DeQuilettes 等[49]基于同样方法得到MAPbI3中I–迁移的EA是(0.19 ±0.05) eV, 最近的研究报告显示, 对于纯碘体系I–迁移的EA在约 0.1—0.6 eV[47,50], 文献中报道的典型EA数据如表1 所列[16,18,21,47,49,50], 可以看出, 尽管材料相同, 所获得的EA数据却差别较大, 造成这样差别的原因是由于计算的模型不同和依据的跳跃方式(迁移媒介)不同, 当然, 所依据的材料状况也有影响, 比如制备过程中引入的富碘和贫碘的情况下, 富碘时I–的EA可低至为约0.2 eV[17,51].但是尽管EA不同, 但它们的趋势是相同的, 即卤素离子比MA+和Pb2+迁移得快.
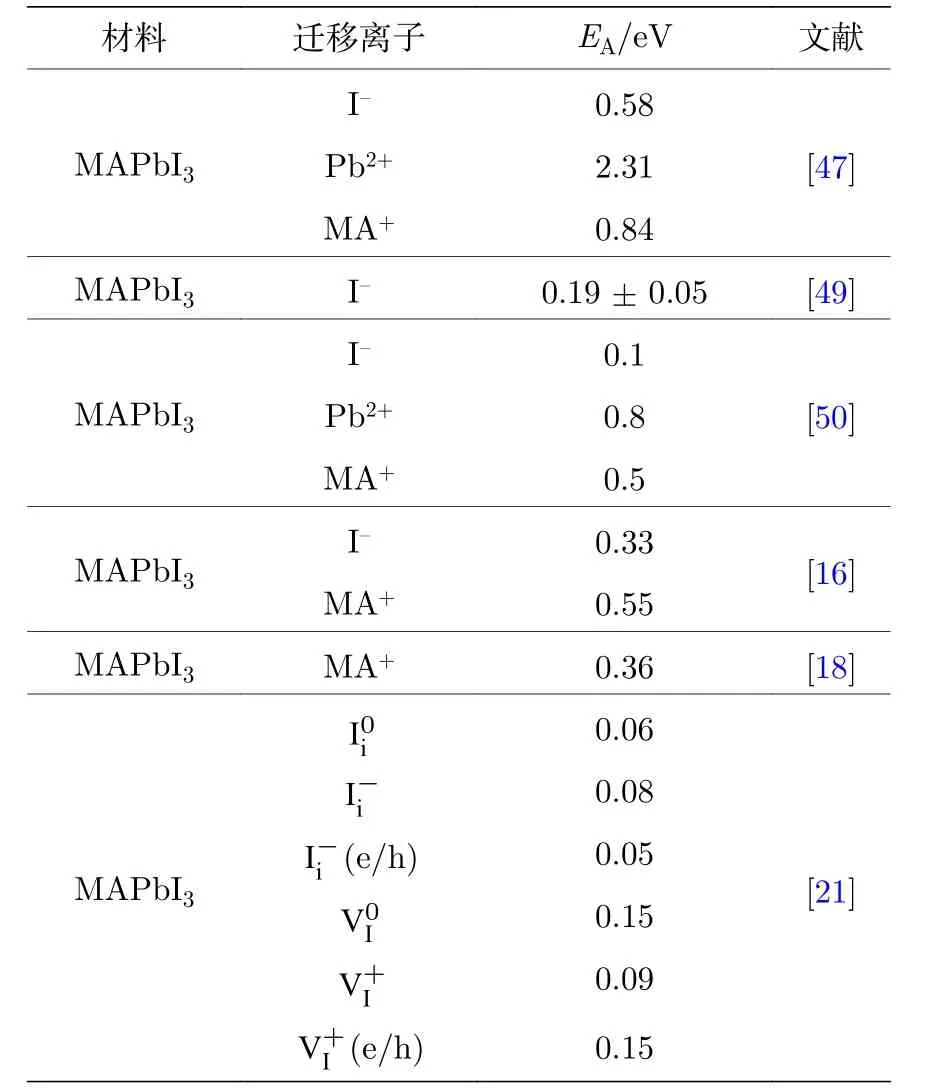
表1 钙钛矿材料的离子活化能Table 1.Ion activation energy of the perovskite material.
致使钙钛矿材料里面存在大量缺陷的原因很多, 从最初合成制备过程引入的缺陷到后期服役过程中新生成的缺陷, 可以概括为以下几方面: 1)溶液法制备的钙钛矿薄膜通常在低温环境下快速结晶制备, 而较低的温度和较快的结晶速度降低了前驱体分子组装成钙钛矿晶体结构的动力, 这和完美单晶的缓慢生长思想形成对比.2)热反应法(比如气相法和气相支持的溶液法)制备钙钛矿薄膜的过程中, 若加热温度过高或加热时间过长, 会使某些组分从晶界处被蒸发掉[23,24].3)在很多情况下,高质量钙钛矿薄膜要求前驱体的比例为非等摩尔量, 比如当MAI 与PbI2的前驱体摩尔比为3∶1时, MAPbI3薄膜的覆盖率最高, 然而此比例偏离了MAPbI3分子结构是所要求的1∶1, 尽管多余的有机组分被认为以气相扩散到大气中, 但开始阶段前驱体的组分不均将在薄膜内引入缺陷.4)理论计算显示钙钛矿材料的分解能垒很低[20,51], 比如MAPbI3的分解能垒仅为0.1 eV, 所以钙钛矿薄膜在服役的过程中很容易发生组分变化.这些使钙钛矿薄膜表面和晶界处存在许多由元素空位和间质元素离子引起较大密度的陷阱.光照条件下, 光激发的电荷会填充陷阱, 扰乱系统从而产生电场, 像常规扩散过程一样, 电场作用下的离子迁移将以钙钛矿薄膜中的缺陷为媒介, 以电场为导向.
最早研究认为体相内的缺陷是离子迁移的主要媒介, 进一步的研究表明表面和晶界也是离子迁移的主要渠道, Xiao 等[25]的研究结果就证明多晶薄膜的晶界处对离子迁移有很大贡献, 除此之外,由于外因引起的晶体结构扭曲也可以产生离子迁移通道[22], Galisteo-Lopez 等[52]利用三维激光共焦断层扫描观察到离子迁移最初阶段所发生的晶体结构扭转.相比较而言, Eames 等[47]的研究比较细致, 他们通过第一性原理的模拟计算, 提出I–沿着八面体的棱边迁移, Pb2+沿着立方晶胞的对角线〈110〉方向迁移, 而MA+必须进入到相邻钙钛矿晶胞的A位置.但是到现在为止, 在热力学层面, 辐照为什么会引起离子迁移以及照射引起的离子迁移的路径机制仍然不清楚, 尽管下一小节所论述的荧光光谱淬灭和增强表明离子移动伴随着缺陷消灭并产生新的复合中心[49,53,54], 但到目前为止, 仍没有直接表征证据来表明这种解释是合理的, 所以关于离子迁移和电池效率的关系仍然没有清晰的结论[46].
配合材料内部极低的缺陷形成能[55], 使得钙钛矿材料的离子迁移发生在很宽的时间尺度(10–3—103s)[56−59], 近年的研究也证实PSCs 呈现出秒量级甚至分钟量级的慢速动力学现象, 这与已经被普遍接受的自由载流子在毫秒时间尺度内发生传输和复合的框架截然不同, 慢动力学过程有望为建立离子迁移和钙钛矿光伏器件的光电转换动力学关系提供新思路.Wu 等[22]指出慢过程可能起源于MAPbI3在电场和/或电荷累积作用下的结构反应, 可能包括MA+的旋转和被置换, 或者晶格畸变, 界面电荷累积在磁滞现象中起了主要作用.但是到目前为止还没有统一的模型可以解释离子和电子导电行为与电池内部电子的传输和复合行为的关系, 因此Pockett 等[56]尝试在经典阻抗等效电路的基础上引入Rionic作为离子迁移过程中的离子复合电阻,Cdl作为钙钛矿/电子传输层或者钙钛矿/空穴传输层界面的离子双电层的电容来修正经典等效电路.Dualeh 等[28]也指出在阻抗分析时,有必要在低频区加一个RC 元素来反映离子电导.
3.2 钙钛矿太阳电池的磁滞现象
PSCs 输出特性的磁滞效应首先被Snaith 课题组[33]报道, 它指的是正向扫描(0→Voc)和反向扫描(Voc→0)的J-V曲线不重合的现象, 这使得真正的J-V曲线很难从常规的输出特性测试获得.前人的研究结果表明, 磁滞效应的大小一方面被测试条件影响, 比如电压扫描速度(1 mV·s–1→100 mV·s–1)[28,33]、光照和外场状态(黑暗中、开路电压或者短路电流)[26,58,59]、扫描方向[26,27,59]; 另一方面被钙钛矿材料本身的特性影响, 比如钙钛矿成分[31,32,58]、晶粒尺寸和电池结构[29,30].磁滞效应给PSCs 的分析和电池的长期稳定性提升都带来了困难.
目前主要用离子迁移行为来解释磁滞现象, 如图4 所示, 在正向扫描过程中, 随着外部电压从0 增大到Voc, 正离子(比如MA+)将扩散到N 区界面, 而负离子(比如I–)将扩散到P 区界面, 离子在界面的堆积完成了对界面的掺杂, 这种自掺杂效应可以扩大两个界面的能带弯曲, 由于与光生电动势方向相反, 在正向扫描时, 将略微抑制光生电荷的提取, 而在反向扫描时, 在界面附近积累的可移动离子会随着外部电场的减小而迁移回钙钛矿材料中, 则前述的自掺杂和能带弯曲对界面电荷收集的影响被解除, 在反向扫描时, 将略微促进光生电荷提取, 正反扫描的叠加就表现出磁滞现象[60], 这与Eames 等[47]和Zhang 等[53]提出的屏蔽场模型很相似.
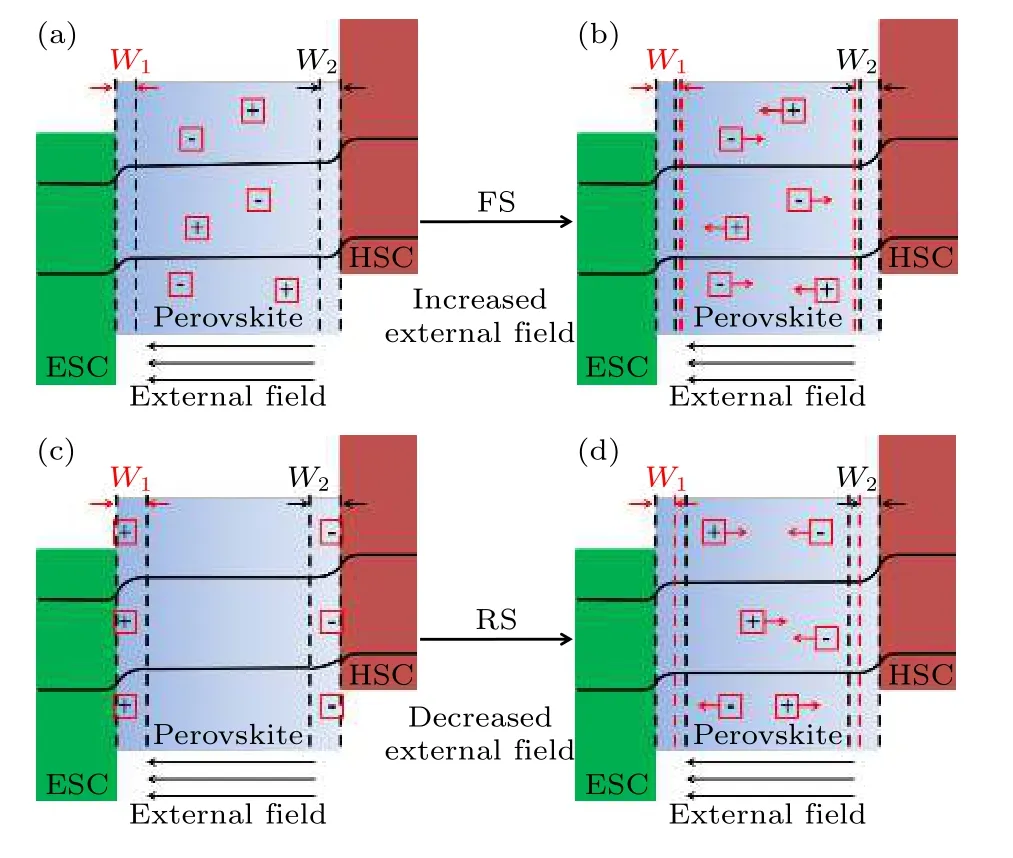
图4 固定电压下PSCs 内部的能级结构对齐关系 (a)和(b)正向扫描; (c)和(d)反向扫描; 其中(a)和(c)不考虑离子迁移行为, (b)和(d)考虑离子迁移行为[60]Fig.4.Schematic illustration of the ion migration: (a) and(b) Electronic band structure alignments of the PSCs at a fixed bias under forward scan; (c) and (d) reverse scan; (a) and(c) without, (b) and (d) with consideration of ion migration[60].
Tress 等[58]也提出了类似的屏蔽场思想, 他们指出离子将聚集在电极的界面处同时屏蔽外加扫描电场对界面电荷提取的作用, 也就是界面电荷的收集效率被偏压控制下的内建电场影响, 且这个修饰过程发生在秒甚至分量级, 使得延迟响应的内建电场更加依赖于电压扫描速度、方向和最初的光电流滞后, 这解释了J-V曲线的磁滞程度强烈依赖于扫描时间和扫描速度的现象.Domanski 等[57]则从实验的角度证明了, 卤素将在1—100 s 的时间尺度聚集到空穴传输层界面, 随着外加偏压的增大, 卤素在空穴传输层界面聚集的现象越严重.除此之外, 他们也指出较大的阳离子迁移时间范围为1000 s, 但所有因离子迁移造成的电池性能差异都会经过一个昼夜的放置后回复到最初性能.
最早Sanchez 等[61]发现, PSCs 载流子复合过程存在持续至秒量级的超慢过程, 第一性原理计算结果显示I–和MA+的迁移是慢速动力学的主要参与者.偏压下的TRPL 结果也佐证负离子的移动和累积是导致数秒至数分钟时间尺度内慢动力学现象的原因[62,63].Li 等[37]通过TPC, TPV 和理论计算表明, 离子迁移主要发生在秒的时间尺度,PSCs 的滞后现象也相应地发生在秒的时间尺度,故从时间维度离子迁移的慢动力学过程可解释为迟滞起源.Unger 等[59]则更明确指出离子迁移中的慢动力学成分是造成磁滞现象的主要原因, 发现使用很快或者很慢的扫描速度都可以得到几乎无磁滞的电池性能, 但只有较慢的扫描速度时, 电池内部才能处于准稳态, 获得更接近于真实的输出性能.此外, 他们用多孔结构(含TiO2多孔层)和面结结构(不含TiO2多孔层)的电池对比, 说明磁滞现象与电池的接触界面也有很大关系.Choi等[36]发现钻进TiO2多孔薄膜内的约70%的MAPbI3晶格结构高度扭曲, 从而离子迁移现象更严重, 这也可以解释介孔结构的电池有更严重的磁滞效应.
与屏蔽场思想不同, Kim 和Park[26]通过一种直接有效的时间分辨光电流法来测试电池的输出特性, 从Si 电池和PSCs 的对比中, 明显看到PSCs光电流反应的拖尾现象, 他们尝试用(3)式来解释正反扫时出现的磁滞现象:
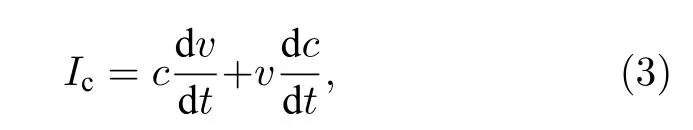
其中,v表示加载到体系中的偏压;c表示偏压下体系的电容; dv/dt表示电压扫描速率; dc/dt表示体系电容随偏压的变化速率;Ic为体系的电容电流,用来维持电池体系到达稳定之前电池内部电容不平衡, 它是一个可以从非稳态电流中加入或者减去的量, 直接影响了输入外电路的电流大小.通常情况下, 正向扫描时Ic是正值, 这种情况下一部分光生电流将以电容的形式储存在电池内部, 相反, 反向扫描时Ic是负值, 电池内部以电容形式储存的电荷尽可能以光生电流的形式输出外电路, 这使得PSCs 正反扫时出现磁滞现象.
综合对比当前的研究进展可以发现用离子迁移形成的屏蔽场解释PSCs 输出特性的磁滞效应应用较为广泛, 但仍有许多亟待探索的领域, 比如离子迁移行为中的慢动力学过程如何从时间尺度上操纵屏蔽场, 以及如何消除或者稳定光电流反应的拖尾现象, 这些对消除或者控制PSCs 的磁滞效应至关重要.
3.3 钙钛矿薄膜的荧光淬灭/增强
随着对PSCs 和钙钛矿薄膜光电性能研究的深入, 研究者逐渐发现了慢动力学过程伴随着钙钛矿薄膜的荧光改变[14,64], 即离子迁移会影响钙钛矿薄膜的荧光特性[9,52].Leitjens 等[65]观察到当外加电场作用在以钙钛矿为中心的多层膜中时, 既可以出现荧光增强又可能出现荧光淬灭.Chen 等[66]则更清晰地指出连续光照将改变钙钛矿薄膜的光电特性, 他们用PL 和TRPL 表征了MAPbI3薄膜的激发特性, 发现在低强度光(0.013 W/cm2)的连续照射下, 钙钛矿薄膜里的可移动离子数量很低,荧光效率有待增强, 当光强增大时(0.25 W/cm2)离子迁移和界面累积过程加剧, 则非辐射复合的加剧使光淬灭.
很多工作尝试用加偏压时的缺陷复合来解释离子迁移和钙钛矿薄膜荧光特性之间的关系[66−69].Sanchez 等[61]观察到PL 的增强和减弱与MAPbI3薄膜的制备方法有关.Jacobs 等[34]等使用TRPL表征了层状结构电极中MAPbI3的慢动力学过程,通过系统改变加载在电极上的电压, 可以观察到一系列可逆的/不可逆的瞬态荧光反应, 并将可逆的瞬态荧光反应归结为薄膜内的陷阱捕获, 而不可逆的瞬态荧光反应归因于离子迁移.Mosconi 等[21]提出光照辐射可以促进碘位空穴和碘离子(Frenkel对)的复合, 这种消去这类缺陷复合中心的方法使得PL 的强度增大.Birkhold 等[54]通过PL/TRPL和扫描开尔文探针显微镜(scanning Kelvin probe microscopy, SKPM)的原位观察, 指出仅偏压引起的MAPbI3自身的离子迁移不一定引入非辐射复合中心, 但是当电荷注入和离子迁移过程联系, 则将产生非辐射复合中心, 且通过密度泛函理论计算发现这个非辐射复合中心是由于Pb2+被还原为Pb0造成的.DeQuilettes 等[49]的研究表明, 光照下钙钛矿薄膜的发光可归因于离子迁移造成的陷阱态密度的数量级降低, 这将改变载流子浓度进而影响光电性能.
另外在混合钙钛矿材料中(比如MAPbBrxI3–x)松散键合的Br–和I–迁移到不同的区域, 在光照条件下MAPbBrxI3–x分开成MAPbI3和MAPbBr3是一种可逆反应, 即光照加快钙钛矿薄膜中Br–和I–迁移并导致卤化物向富碘和富溴结构域的偏析生成MAPbI3和MAPbBr3, 但当材料在黑暗中储存时仍可回复最初状态.在表征的过程中可观察到电致发光峰随着电偏压的增大而分裂为两个峰值, 出现了偏聚后的新相, 这种现象后来被命名为“Hoke 效应”, 类似的光诱导的相偏析也被其他研究人员证实[48].
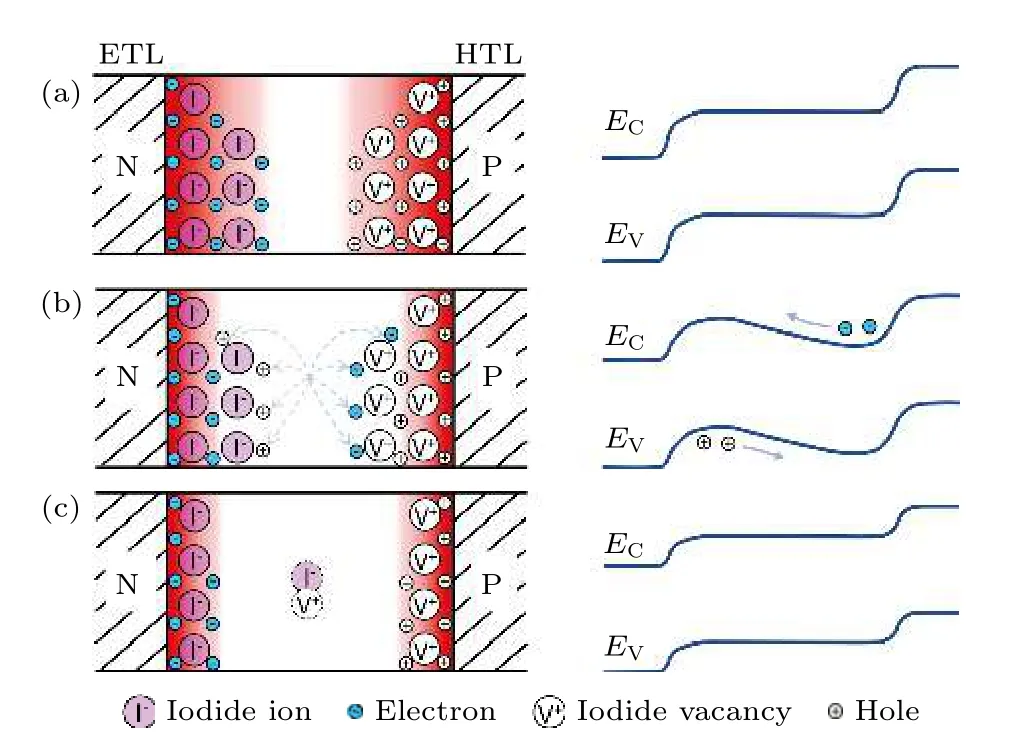
图5 离子和电子分布示意图(左侧)和相应的能级分布(右侧) (a)黑暗中; (b)光照的瞬间; (c)光辐照一段时间后; 其中, 红色阴影区域为耗尽区域, 阴影等级表示电场强度, 由于光照下产生的内建电场, 图(b)和(c)中耗尽区域宽度逐渐减小, 在图(b)中, 左侧的虚线箭头代表离子迁移到平衡状态之前, 电子和空穴在萎缩的耗尽层区域的重新分布, 与此同时能带的弯曲减少了对外的静电流[70]Fig.5.Schematics of ionic and electronic carrier distributions (left) and corresponding band diagrams (right) for three situations of interest: (a) Dark equilibrium; (b) immediately after light turns on; (c) after prolonged illumination.Where the red-color shaded region is the depletion region with the shade grading indicating the electric field strength,note that the depletion region width reduces in panel (b)and (c), because of photovoltage bulid-up after illumination;in panel (b), dashed arrows on the left indicate redistribution of electrons and holes upon the shrinkage of the depletion region but before ions move to new equilibriums, while distortion of the band diagram on the right results in a reduction in the net currents[70].
澳大利亚新南威尔士大学的Lan 以经典半导体物理学为基础, 提出了一种更为简单的离子迁移和钙钛矿薄膜光电性能相关联的独特模型.Lan等[70]基于经典太阳电池物理对黑暗下、刚开始光照和光照一段时间后PSCs 中的离子和电子分布进行分析, 如图5 所示, 指出在PSCs 中耗尽层的宽度从钙钛矿薄膜分别与电子传输层和空穴传输层的接触面开始向钙钛矿层体相延伸约100 nm[55],这表明暗态下电池内部的离子偏置现象很严重, 但体系内的离子场和电子场都处于平衡状态, 所以体系内的能级分布可均匀贯穿两侧的结区和中间的体区.当把PSCs 放置到光下的瞬间N 和P 两侧的耗尽层宽度变窄, 在体系内的离子迁移稳定平衡的同时体系内的电子也高速迁移以抑制离子场的作用, 但电子扩散的方向与外接电路的电子迁移的方向相反, 由于电子的屏蔽作用离子迁移的速度很慢, 体系的能级关系不恒定, 但存在最低值.随着光照时间的延长, 体系中混合的离子-电子系统仍将达到新的平衡状态, 其中的离子场和电子场分布如图5 所示, 体系的能级分布也处于平衡状态, 但其中的电子漂移影响了外部电池性能的测试结果.Lan 等[70]以正向扫描为例, 提出在扫描的过程中随着扫描偏压的减小, 耗尽层逐渐变窄, 滞后的带电离子会扰乱钙钛矿的带隙结构, 这造成了体系回复平衡状态之前静电流的减少, 和反扫的曲线相比就出现了磁滞现象.除此之外, 离子迁移行为将影响电荷和空穴的分布, 进一步同时影响缺陷复合(shockley-read-hall)和俄歇过程(Auger process),所以会不同程度地影响辐射复合和非辐射复合的比例.Lan 等[70]也指出非辐射复合由直接缺陷复合和俄歇复合两部分组成, 所以表征的PL 结果将由缺陷复合和俄歇复合的结果决定, 但实际上从非平衡状态到平衡状态的整个过程中, 钙钛矿材料的质量和测试环境等因素让两种复合之间的大小无法确定, 所以会出现随着光照时间不同, 非辐射复合有可能不变、增强或减小这3 种趋势.但无论是否对非辐射复合产生影响, 这个太阳电池物理过程将解释随着辐照时间的延长电池性能的不稳定变化.
3.4 钙钛矿太阳电池的失效
目前, 大多数PSCs 在环境条件下, 连续光照时将迅速失效.Miyano 等[71]表明由于离子迁移行为也是导致电池失效的原因之一.在钙钛矿薄膜两侧加上接触电极后, 研究者发现在加载偏压后, 离子迁移的结果甚至会造成钙钛矿材料的降解[25,35].Leijtens 等[35]也证实, 环境因素耦合外加电场对将更容易促进MA+的迁移, 导致钙钛矿结构不可逆地退化成PbI2.但其实根据迁移离子的来源: 1)之前钙钛矿薄膜中已经存在的离子; 2)分解后的钙钛矿材料提供[18,34], 离子迁移和钙钛矿材料失效应该呈协同恶化的关系, 也相当于光激发使钙钛矿材料发生降解, 将促进可移动碘离子的量.Di Girolamo等[72]使用原位检测技术, 完全模拟电池工作环境,探讨了离子迁移引起的电池失效的原因, 他们指出足够的电荷注入使离子迁移, 这引起钙钛矿材料的无定型化和卤素偏聚,Voc的升高是由于形成空间电荷区, 钙钛矿材料的自掺杂效应引起, 但同时卤素的偏聚以及钙钛矿的无定型化, 将会阻止电荷的收集, 引起Jsc和FF 降低.
除此之外, 钙钛矿材料易吸附多类化学物质,比如潮湿[73]和有机极性分子[74], 这些物质在光下将与钙钛矿材料发生光化学反应致使PSCs 失效.Aristidou 等[75]提出MAPbI3结构中的碘缺陷是超氧物形成的关键部位, 在光照射下超氧的强还原性将使钙钛矿材料分解.Ahn 等[74]表明钙钛矿材料对电荷的捕获对降解机制起决定性作用, 比如有机阳离子通过俘获电荷从而诱导局部电场去质子化.Bryant 等[13]证明了光和氧诱导的降解比更广泛研究的水分诱导降解更快, 是MAPbI3暴露于阳光下降解的主要原因.Abdelmageed 等[76]利用X 射线衍射和超小波研究了MAPbI3材料和相应器件在低湿环境中的光诱导结构降解机理, 他们发现滤除紫外线可以显著提高PSCs 的稳定性.
但实际上, 离子迁移和光氧化有可能是一个同时进行的过程, 其中各个功能层的接触界面是PSCs 稳定性的薄弱点[63], Jung 等[62]等用原位透射电子显微镜观察了选择性接触界面在电场下的不稳定性, 直接观察到了正向偏压下氧分子从TiO2到MAPbI3的扩散[77], 同时界面结构和化学降解也在Spiro-OMeTAD(2,2',7,7'-Tetrakis[N,Ndi(4-methoxyphenyl)amino]-9,9'-spirobifluorene)/MAPbI3界面观察到, 即碘元素与Spiro-OMeTAD之间的相互作用[78], 更多的研究结果也以PbI2产物的形成来证实MAPbI3的成分分解[79,80].
通过封装隔绝环境中引起不稳定的因素后,2016 年Bryant 等[13]以MAPbI3为研究对象, 对比了空气和N2氛围下薄膜的稳定性, 结果表明空气环境下的电池在不到4 h 时间性能降低了80%,相反N2氛围下电池在12 h 的连续测试中性能持续缓慢提高, 他们认为空气中的O2是引起钙钛矿薄膜失效的关键因素.Dong 等[81]比较了封装后的电池稳定性, 他们发现封装后以TiO2为电子传输层的PSCs 在户外性能可以稳定432 h, 可见封装有助于提高电池稳定性, 然后针对PSCs 户外稳定服役25 年的要求, 仅仅针对封装材料以及封装工艺本身也需要进行不断探索, 且封装并不能解决电池内部的离子迁移现象.
3.5 离子迁移的实验证据
由于离子迁移过程发生在原子尺度, 对这一过程的表征已被证明是一项艰巨的任务.DeQuilettes等[49]使用共聚焦荧光显微镜和化学成像相结合,通过飞秒二次离子质谱(time-of-flight secondaryion-mass spectrometry, ToF-SIMS)对MAPbI3深度方向的研究来直接呈现碘离子的迁移证据.他们首先选取了MAPbI3薄膜二维荧光照片中的一个暗点, 对其使用脉冲激发光光曝几分钟, 同时记录典型的荧光激发行为的增强, 如图6(a).然后在同一个区域用ToF-SIMS 表征薄膜深度方向的碘元素信号如图6(b)所示, 图6(c)呈现了图6(b)中划线区域碘元素分布的柱状图结果, 从结果可以看出, 在光照条件下PL 的寿命和强度随着时间的推移而显著增加, 使用F-SIMS 深度剖面来表明PL 的上升与薄膜中碘的重新分布有关, 从而为光诱导的卤化物迁移提供了有力的证据.Domanski等[57]也通过ToF-SIMS 观察到了电场作用下钙钛矿材料内部明显的I 元素和Br 元素偏聚的状态.
Birkhold 等[54]使用原位荧光配合SKPM 分析了以钙钛矿材料为核心的层状结构电极在电场下的离子和电子运动行为(图7), SKPM 对探测电场驱动下层状电极结构中的离子迁移非常有用, 一般情况下SKPM 测试电极和样品之间的接触电势差, 而缺陷和电荷分布的改变会影响接触电势差,所以可通过SKPM 来分析层状结构电极里的离子迁移.他们选用了加与不加绝缘层的两种样品, 两种样品的区别在于绝缘层的加入将阻挡界面电荷的提取和传输.当样品界面处不加绝缘层时, 样品断面厚度范围内压降大体上与所加偏压一致, 偏压撤掉后, 仅负极区域有高电势残留, 相反对于加上了绝缘层的样品, 样片断面厚度范围内压降的分布截然不同, 存在正负两个电势中心, 去掉偏压后,正负两个电势中心依然保留, 所以在薄膜的两侧形成了两个像势垒一样的电荷屏障.而造成SKPM测试结果发生变化的原因是由于绝缘层的加入阻挡了电子对薄膜的注入, 以及薄膜内离子向外界的迁移, 所以积攒到两侧的离子影响了SKPM 测试的接触电势差.
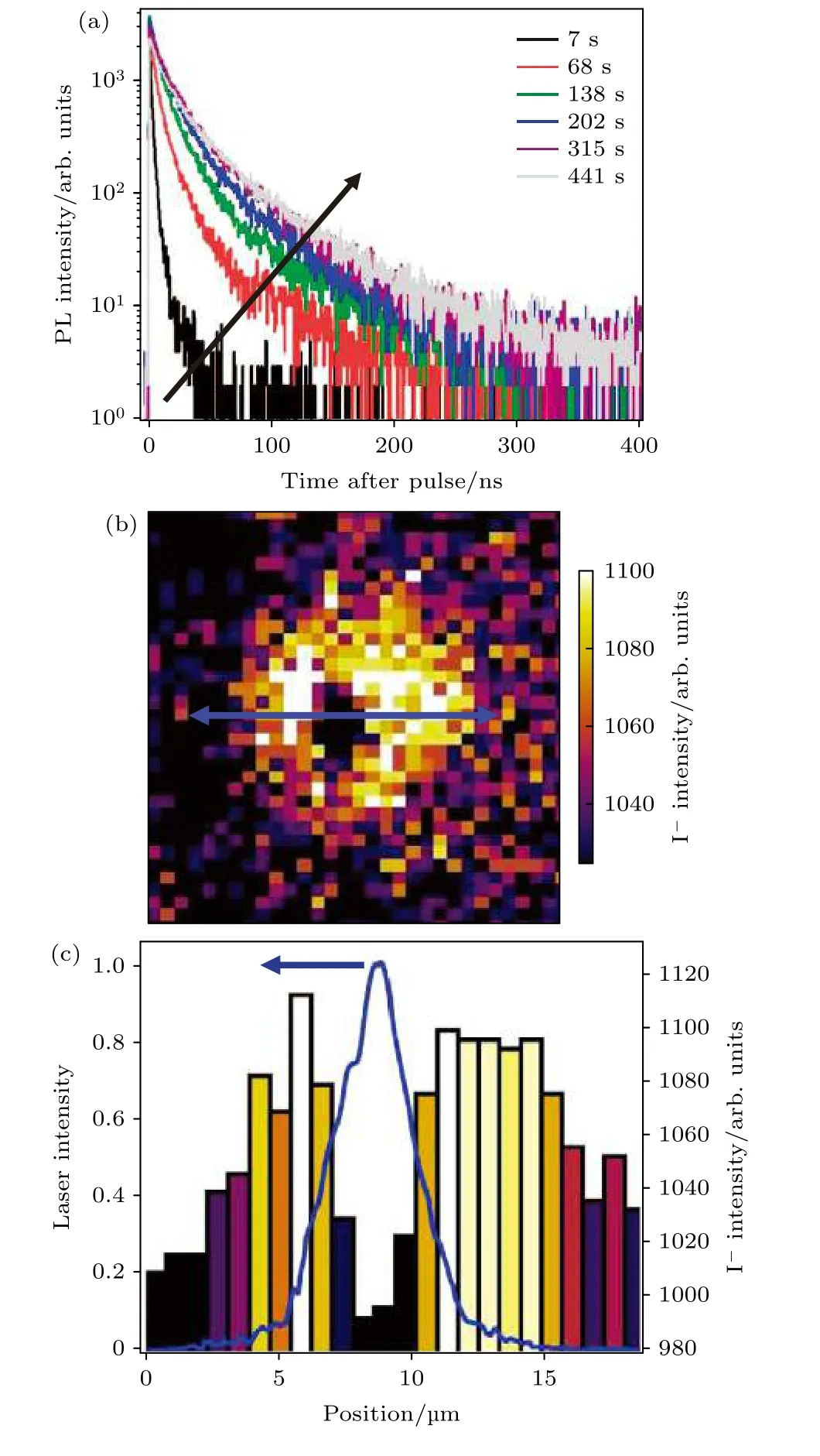
图6 光曝后碘的重新分布现象 (a)不同光曝时间下MAPbI3 薄膜的瞬态荧光淬灭结果, 其中脉冲激发光源为470 nm, 1.2 kJ/cm2; (b) ToF-SIMS 采集的钙钛矿薄膜内碘元素在深度方向的信息, 标尺为10 µm; (c)是对图(b)中蓝线区域碘分布的线扫描结果(右轴), 照明激光的空间轮廓测试结果被显示在左轴[49]Fig.6.Iodide redistribution after light soaking: (a) A series of time resolved photoluminescence decays from a MAPbI3 film measured over time under illumination before ToFSIMS measurements, and the sample was photoexcited with pulsed excitation (470 nm, 1.2 kJ/cm2); (b) ToF-SIMS image of the iodide (I–) distribution summed through the film depth (the image has been adjusted to show maximum contrast), scale bar, 10 µm; (c) line scan of the blue arrow in panel (b) to show the iodide distribution (right axis), where the measured spatial profile of the illumination laser (blue)is shown on the left axis[49].
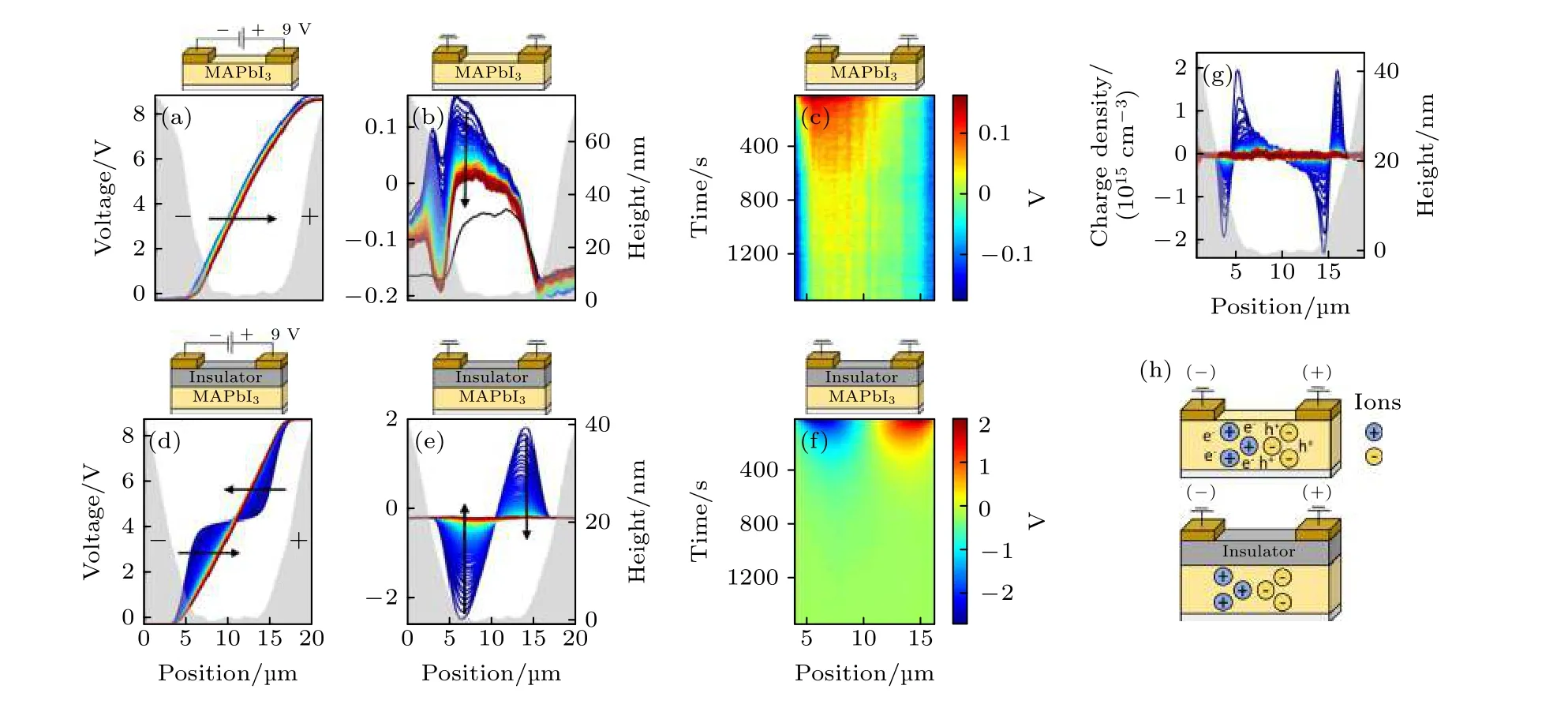
图7 SKPM 在MAPbI3/Au (a)−(c)和MAPbI3/PMMA/SiO2/Au (d)−(f)电极界面的单线扫描结果 (a)和(d)从左侧为两类样品加上+9 V 的偏压; (b), (c), (e)和(f)是关掉+9 V 正向偏压后接地; (g)是去掉+9 V 偏压后MAPbI3/PMMA/SiO2/Au 样品中的电荷密度分布; (h)是去掉偏压后, 两类样品中电子和离子的分布示意图[54]Fig.7.SKPM scan of a single line within the electrode gap of (a)−(c) MAPbI3/Au and (d)−(f) MAPbI3/PMMA/SiO2/Au, measured (a), (d) with a +9 V bias applied to the right electrode and (b), (c), (e), (f) at 0 V bias after turning off the +9 V bias; the black line in (b) displays the SKPM CPD signal prior to biasing; (g) charge density in MAPbI3/PMMA/SiO2/Au after bias; (h) illustration of electronic and ionic charge distribution after electric biasing[54].
Shao 等[29]也借助SKPM 技术直接观测到了I–的迁移.之前理论已经预测了I–比MA+和Pb2+更容易迁移, 但MA+的迁移很难被直接观测到.最新的光热诱导共振显微术(photothermal induced resonance microscopy, PTIR)可以直接测量MA+的分布, 该技术联合了傅里叶变换红外光谱的化学元素检测能力和原子力显微镜的呈像能力.Yuan等[18]借助该技术观察了层状MAPbI3太阳电池电极化之后的MA+离子分布, 研究发现, 通过加载一个1.6 V/µm 的电场100 s, MA+离子离开阳极和中间区域, 累积在阴极区.Yang 等[19]将电池在10 nA 的直流电下50 ℃服役1 周, 采用常规X 射线衍射(X-ray diffraction, XRD)和能谱方法(Xray enery-dispersion spectroscopy, EDS)测试离子迁移情况, 如图8 所示.
3.6 离子迁移的抑制方法
离子迁移可以改变电荷注入速率以及电子和空穴的复合行为, 对PSCs 的磁滞现象、钙钛矿薄膜的荧光淬灭/增强以及电池失效都有直接影响,因此对离子迁移的抑制一直是研究热点, 钙钛矿薄膜的体相和/或界面钝化、钙钛矿薄膜的组分调控和钙钛矿晶体的维度调控都是有效的抑制方法.
钙钛矿薄膜的体相和/或界面钝化主要目的在于钝化体相和/或界面缺陷, 阻断离子迁移的路径,具体的实施途径包括: 1)向前驱体溶液中添加钝化剂, 随着钙钛矿前驱体的结晶析出, 所添加的钝化剂作用于钙钛矿晶体体相和/或界面的缺陷.例如, Deng 等[82]在钙钛矿溶液中添加极微量的表面活性剂L-α-磷脂酰胆碱(L-α-Phosphatidylcholine),一方面钝化了钙钛矿薄膜中的Schottky 缺陷, 一方面抑制钙钛矿薄膜/空穴传输层界面的缺陷.Wu 等[83]采用2, 3, 5, 6-四氟-7, 7, 8, 8-四氰基喹二甲烷(2, 3, 5, 6-tetrafluoro-7, 7, 8, 8-tetra-cyanoquinodimethane, F4T CNQ)钝化钙钛矿薄膜,F4T CNQ 是一种强的电子抽吸分子, 促进了界面电荷的提取, 他们将添加剂工程和溶剂退火工艺相结合, 得到了表面全覆盖的优质MAPbI3薄膜,PSCs 的效率超过20.0%, 几乎没有磁滞现象.实验结果表明富勒烯也可被加入到钙钛矿前驱体溶液中, 钝化钙钛矿薄膜内部的缺陷[84].2)使用与前驱体组分类似的盐类替换前驱体的某一成分, 通过参与钙钛矿结晶过程钝化钙钛矿薄膜的缺陷.例如,Aristidou 等[75]采用碘盐(phenylethylammonium iodide, MAI or Trimethylsulfonium iodide, (碘化苯乙基胺、碘化甲胺或碘化三甲基磺胺))钝化钙钛矿薄膜, 显著提高了PSCs 的稳定性, 研究表明MAPbI3结构中的碘缺陷是超氧物形成的关键部位, 碘盐处理可以减少碘缺陷, 减少超氧物的形成,从而增强薄膜和器件的稳定性.Buim 等[51]使用醋酸铅作为前驱体, 一方面提供了贫碘的环境, 一方面提高了电荷扩散长度.3)使用钝化剂处理钙钛矿/电子(空穴)传输层界面, 从而钝化钙钛矿活性层与其他功能层界面处的缺陷.例如, Bi 等[85]采用脂肪族荧光两亲性添加剂1, 1, 1-三氟碘化铵(1, 1, 1-trifluoro-ethylammonium iodide)修饰钙钛矿和空穴传输界面, 优化后的PSCs 磁滞现象可以忽略不计.因为富勒烯可以扩散进晶界, 钝化钙钛矿薄膜表面的缺陷, 因此其常作为电子阻挡层应用于反向PSCs, 以消除电池的磁滞现象[86].除此之外, 由于“流动离子”主要沿晶界迁移, 制备大晶粒钙钛矿薄膜是抑制离子迁移的可行方法, 因此,增大钙钛矿薄膜的晶粒尺寸可有效抑制PSCs 的磁滞现象[26], Ke 等[30]在钙钛矿前驱体中添加Pb(SCN)2, 将钙钛矿薄膜的晶粒尺寸从100—400 nm 增大到2 µm, 明显抑制了电池的磁滞现象.
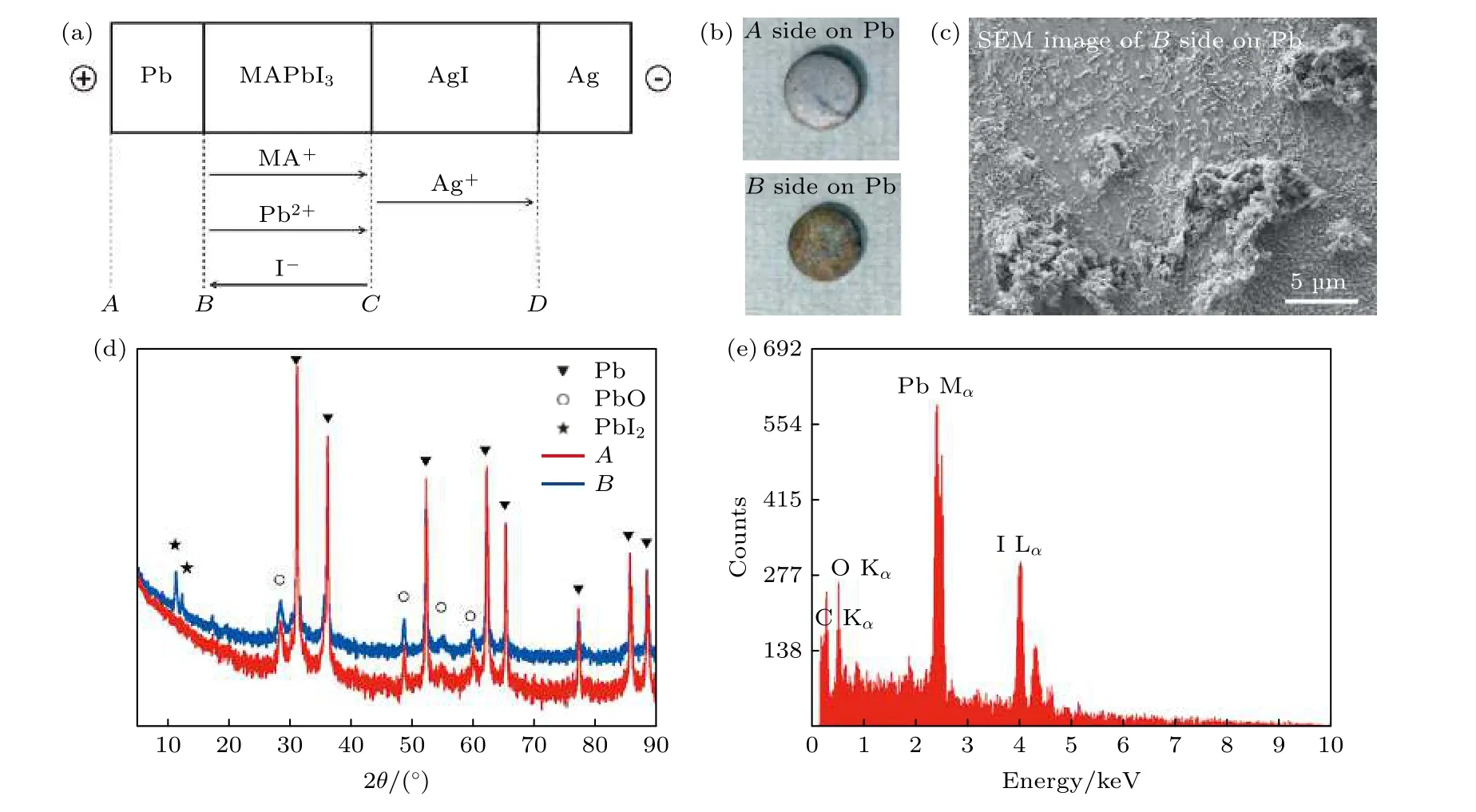
图8 在Pb/MAPbI3/AgI/Ag 电池中(a)电场作用下带电离子的流动方向; (b)电池A, B 面的图片; (c) B 面的SEM 图片; (d)在10 nA 直流电下服役1 周后A, B 两面的XRD; (e) B 面Pb 元素的EDS[19]Fig.8.(a) Flow directions of the charged ion species in a Pb/MAPbI3/AgI/Ag cell under electrical bias; (b) images for surfaces A and B; (c) SEM image of surface B on the Pb pellet; (d) XRD patterns of surfaces A and B of the Pb disk after applying a direct current of 10 nA for a week; (e) EDS spectrum for surface B of Pb[19].
组分调控也被认为是一种行之有效的抑制离子迁移的方法, Haruyama 等[16]认为与MAPbI3相比, FAPbI3中I–和MA+的EA均增加, 提出了利用大有机阳离子抑制钙钛矿离子迁移的可能性.Jeon 等[32]将MAPbI3添加到FAPbI3中, 减小了PSCs 的磁滞.Ahn 等[74]开发了一种混合的MA0.6FA0.4PbI2.9Br0.1钙钛矿, 确保了PSCs 的高性能和稳定性.DFT 模拟计算出的EA值表明混合MAPbBrxI3–x钙钛矿中, Br 可以稳定基体并阻止离子迁移[60].Yang 等[87]认为混合卤素离子的钙钛矿配方中, 若在晶界引入Cl–后, 工艺可以得到增强, PbI2自钝化钙钛矿也是一种提高光伏性能和减少迟滞的有效方法.近期强大的温度相关瞬态技术已允许测量材料的EA, 所以EA的获得被用于评估和预测多组分钙钛矿材料的离子迁移能力[88].
以更大的阳离子来替换MA+, 甚至改变3D钙钛矿的结构单元也是研究者的努力方向.Chen等[89]证实离子迁移在二维PEA2PbI4钙钛矿中被大大抑制.Wang 等[90]将正丁基铵离子引入到混合阳离子铅卤钙钛矿, 在三维钙钛矿晶粒之间可以观察到“板状”2D 相晶体, 由于界面晶界钝化, 在这种“异质结构”膜中可显著观察到抑制非辐射复合的作用, 借助二维和三维钙钛矿相之间的工程异质结构, 继而提高PSCs 的稳定性.Lee 等[91]制备帽状二维钙钛矿以保护三维钙钛矿不受水分的影响, 同时抑制离子迁移.Xiao 等[92]进一步证实了二维钙钛矿中VI–和VMA的形成能增大, 与它们的三维类似物相比, 进一步支持了二维钙钛矿中离子迁移被抑制.
4 TiO2/钙钛矿界面的紫外不稳定现象、抑制办法和钝化效果的实验证据
4.1 紫外不稳定现象
高效n-i-p 结构PSCs 常使用钙钛矿薄膜和TiO2作为光吸收层和电子传输层, 但在长期光照后可以发现TiO2/钙钛矿界面损坏严重, 这也是电池性能衰退甚至失效的一个重要原因.2015 年日本兵库县立大学的Umeyama 等[93]在全光谱下以单独的TiO2/钙钛矿双层膜为研究对象, 提出“光”、“TiO2”和“空气”三者联合将使钙钛矿薄膜降解.稍后日本大阪技术研究所的Mosconi 等[94]以整个PSCs 为研究对象, 发现当钙钛矿与其他功能层形成界面时, 电荷在界面传递的过程会加速钙钛矿的降解.2016 年韩国高丽大学的Lee 等[95]更深入地揭示了太阳光谱中的紫外光将通过TiO2表面丰富的缺陷态, 在TiO2的禁带内形成亚稳态深能级缺陷, 它将从钙钛矿结构中夺取卤素负离子的电子, 进而使钙钛矿降解[96].2018 年德国卡尔斯鲁厄理工学院的Farooq 等[97]证明了入射紫外波长越短, 钙钛矿被降解的越严重.这些都充分显示尽管TiO2/钙钛矿界面在短时间内可促进PSCs 的高效, 但它并不是一个稳定界面.
根据TiO2是一种优异光催化材料的事实, 不难明确其对TiO2/钙钛矿有严重破坏作用的原因:1) TiO2对紫外线非常敏感, 其吸收范围恰在紫外光区; 2) TiO2颗粒表面有丰富的氧空位, 这些为降解相邻材料提供了大量的活性位点.所以, 在太阳光中的紫外光照射下, TiO2表面丰富的缺陷(Ti3+)被激活, 这些局域捕获状态(Ti3+)的电子将与O2分子结合, 生成复杂的Ti4+-O2–, TiO2被紫外光激发所产生的价带空穴将与Ti4+-O2–处的电子复合, 从而解吸所吸附的O2分子, 并留下带有正电荷(Ti4+)的氧空位.空穴传输层中多余的空穴将也与TiO2导带中剩余的未结合的电子复合.带正电荷的氧空位(Ti4+)是位于TiO2材料导带以下的深能级缺陷, 倾向于从卤素负离子中提取电子, 从而破坏钙钛矿材料的电平衡, 以至于PSC 的结构从TiO2/钙钛矿界面分解, 其原理如图9 所示[98].由于O2分子的缓慢和可逆的解吸速率, 深能级缺陷区域内的O2分子的分压决定着具有催化活性的氧空位的数量, 但在TiO2材料的表面O2的扩散逐渐被紫外光加速, 因此, TiO2催化降解钙钛矿的效果非常显著, 通常在1000 W/m2光照下,PSCs 在几分钟至几小时内效率明显降低甚至失效.所以, 实现TiO2/钙钛矿界面紫外稳定化也是重要的研究方向.
4.2 抑制办法
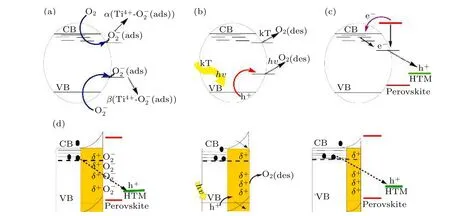
图9 紫外线照射下TiO2 材料的光催化(a)−(d)行为及机理(TiO2 材料中存在大量缺陷, 通过吸附和脱附O2 分子的过程, 形成深能级缺陷Ti4+, 它通过从卤素负离子中提取电子的方式破坏了钙钛矿结构的电平衡)[98]Fig.9.Photocatalysis of TiO2 material under UV illumination: (a)−(d) there are abundant defects in TiO2 material.During the absorption and deabsorption of the O2 molecular, the positive charge (Ti4+) is formed, which will extract electrons from halogen negative ions, thus destroying the electrical balance of perovskite structure[98].
最早报道的是从入射光出发, 在电池中添加下转换荧光材料, 将入射的紫外光转换成可见光, 即将高能量的紫外光子转换成更多的可见光子, 这既避免了紫外损害又增强了主要用于光伏转换的可见光强度.Jin 等[99]将YVO4:荧光颗粒加入TiO2电子传输层中; Wang 等[100]将CsPbCl3:Mn 量子点沉积到PSCs 的入射光一侧, 由于该材料与入射紫外光的波长范围和强度的匹配度良好,所以电池的紫外稳定性明显提升.但是由于下转换的原理是基于稀土元素的f-f 或f-d 跃迁, 所以很难实现对全部波段的紫外光进行转换.
也有一部分研究工作尝试替换TiO2为无光催化活性的材料, 比如You 等[101]使用p 型NiOx和n 型ZnO 纳米粒子薄膜作为空穴层和电子传输层,尽管显著提高了稳定性, 但ZnO 又使PSCs 的磁滞现象加剧.Carnie 等[102]则使用绝缘的Al2O3纳米粒子直接添加到钙钛矿前驱体溶液中, 但作为电池的支撑层, Al2O3的带隙与钙钛矿并不匹配, 尽管有报道显示可提高Voc, 但对Jsc的贡献很有限.相比较而言, SnO2具有较宽的带隙、更高的导电性和更强的电子传输能力强, 是一种研究相对广泛的电子传输层材料.Wang 等[103]通过原子层沉积制备了SnO2电子传输层, 但PSCs 的最高效率仅为18%.Jiang 等[104]通过简单的溶液法一步制备了3—4 nm 的SnO2晶粒, 并利用旋涂法堆垛成膜, 这与原子层沉积相比有效降低了SnO2电子传输层的制备成本.Wang 等[105]以硫元素钝化SnO2电子选择层, 试图减轻界面复合提高以SnO2为电子传输层PSCs 的效率.Hu 等[106]将SnO2插入TiO2/钙钛矿界面中间, 因为界面缺陷被抑制和界面能量损失被减少, PSCs 的电池的最优效率达到20.50%.
另一类是使用化学性质稳定的薄膜在不影响界面电荷传递的同时, 钝化TiO2表面的缺陷.You 等[101]采用生物聚合物肝素钠(biopolymer heparin sodium, HS)中间层锚定MAPbI3/TiO2界面, HS 的存在有效地抑制了太阳能电池的迟滞现象, 使PCE 提高了20.1%.Sidhik 等[107]采用石墨烯纳米片(graphene nanoplatelets, GnPs)钝化了TiO2表面缺陷, PSCs 甚至表现出可忽略的迟滞现象.Shih 等[108]采用多种氨基酸钝化TiO2电子传输层, 同时调控了钙钛矿晶体在界面上的取向, 促进了电荷传递.Ogomi 等[109]在TiO2/钙钛矿界面之间插入PEROVI3(HOCO-R-NH3+I monolayer), 通过PEROVI3晶体的生长调控来得到最优钝化效果.
理想的钝化膜要求其极薄且全覆盖在TiO2薄膜的内外表面, 然而在微观尺度实现钝化膜对不足1 µm 的TiO2薄膜的极薄且全覆盖对沉积工艺和成膜行为控制提出了严苛要求.必须要提的是,不仅仅在PSCs 中, 在量子点敏化太阳电池、染料敏化太阳电池和硅太阳电池中, 为了阻止界面电荷复合, 也同样需要极薄且全覆盖的钝化膜[110,111],且也存在理想钝化膜难实现的技术问题.目前比较有效的沉积工艺包括: 1)原子层沉积(atomic layer deposition, ALD), 例如, 瑞士联邦理工学院的Lee 等[112]以Al(CH3)3和H2O 为前驱体, 在TiO2薄膜表面沉积了1—5 nm 厚的Al2O3薄膜, 但是ALD 技术复杂的工艺流程和高成本限制了其工业化应用.2)单层自组装(self-assembled monolayer,SAM), 例如, 牛津大学的Snaith 等[113]以化学结构中含有羧基和苯环的苯甲酸为中间体, 利用羧基与TiO2的桥接以及苯环与C60的相容性, 通过提拉浸泡过程实现了C60在TiO2表面的单层自发组装, 但是SAM 技术只适合于有机分子的组装, 而有机物存在老化和被TiO2催化降解的问题.3)分步离子层吸附反应(sequential ionic layer adsorption and reaction, SILAR), 例如, 韩国浦项科技大学的Hwang 等[114]首先将TiO2薄膜浸泡在CdSO4溶液中物理担载上Zn2+, 然后再将TiO2薄膜浸泡到Na2S·9 H2O 溶液中, Cd2+与S2–在TiO2薄膜表面反应得到几个纳米厚的CdS 沉淀, 但由于Zn2+在TiO2表面担载量完全依靠于浸泡过程的随机分布, 所以不易实现均匀且全覆盖的钝化膜.4)一步溶液法(one-step solution method)[115], 与其他方法相比, 其低成本、易操作且适合廉价、小分子尺寸、性质稳定的无机盐薄膜的制备.2014 年清华大学的Wang[116]等率先将CsCO3水溶液滴加到TiO2薄膜上, 经过旋涂并干燥, 制备了CsCO3覆盖的TiO2薄膜, 将PSCs 的效率提高了20%, 但由于CsCO3前驱体结晶行为不可控, 以及后期钙钛矿前驱体溶液对CsCO3薄膜的严重溶解, 使CsCO3薄膜的覆盖率很低, 甚至无法在透射电子显微镜下被找到.2016 牛津大学的Li 等[117]选用在钙钛矿前驱体溶液中溶解度较小的CsBr, 通过一步溶液法制备钝化膜, 但由于CsBr 前驱体的结晶行为不可控及后期被钙钛矿溶液溶解的问题仍未得到彻底解决, 所以CsBr 薄膜呈团簇状分布在TiO2薄膜上, 但即便不全覆盖, PSCs 的连续光照稳定性却明显提高.再到2018 年瑞士洛桑工业技术学院的Seo 课题组[118]跳出微液膜内溶质结晶行为不可控的限制, 将CsBr 溶解在TiO2悬浮液中,一步沉积CsBr 和TiO2混合的薄膜, 但这种方法虽然促进或实现了全覆盖, 却增加了电池内部的传输电阻.另外, PbI2, CuI 和SbI3等都是一步溶液法常用的钝化材料.
4.3 钝化效果的实验证据
常规研究大多通过外量子效率、电化学阻抗谱以及PL/TRPL 等测试来表明钝化膜在提高电池稳定性方面的有用性, 但由于没有直接且有效的表征手段, 对深层次的钝化膜作用机理还不能给出有效解释.目前已报道的建立钝化行为与TiO2表面缺陷态浓度关系的表征方法少之又少, 日本九州工业大学的Kim 等[26]使用“Thermally stimulated current method”得到TiO2在–4.2 eV 的导带位置经过SbI3钝化后, 缺陷态浓度从1016/cm3降低到1014/cm3, 但这种方法并不能直接反映TiO2禁带以内的深能级缺陷.所以, 采用更直接的手段表征TiO2薄膜内深能级缺陷的类型以及浓度是明确钝化效果的主要瓶颈.
目前文献中常使用热导纳谱(thermal admittance spectroscopy, TAS)的分析结果来反映钝化前后界面缺陷态浓度的变化.前文中提到Snaith课题组的Li 等[117]用电化学工作站测试了PSCs的TAS, 如图10 所示.TAS 是一种描述浅层和深层缺陷的有效技术, 已广泛应用于有机太阳电池陷阱态密度的能量分布(t-DOS), 由角频率相关电容导出的方程为
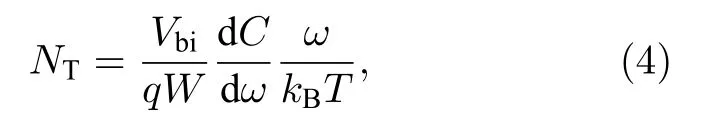
其中C为电容,ω为角频率,q为基本电荷,kB为Boltzmann 常数,T为温度.Vbi和W为内建电场和耗尽宽度, 可从Mott-Schottky 曲线中获得.角频率ω定义了能量分界:
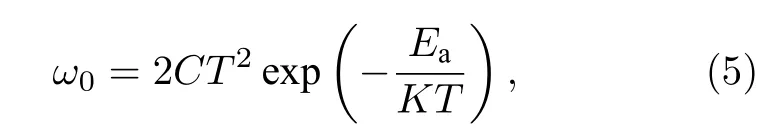
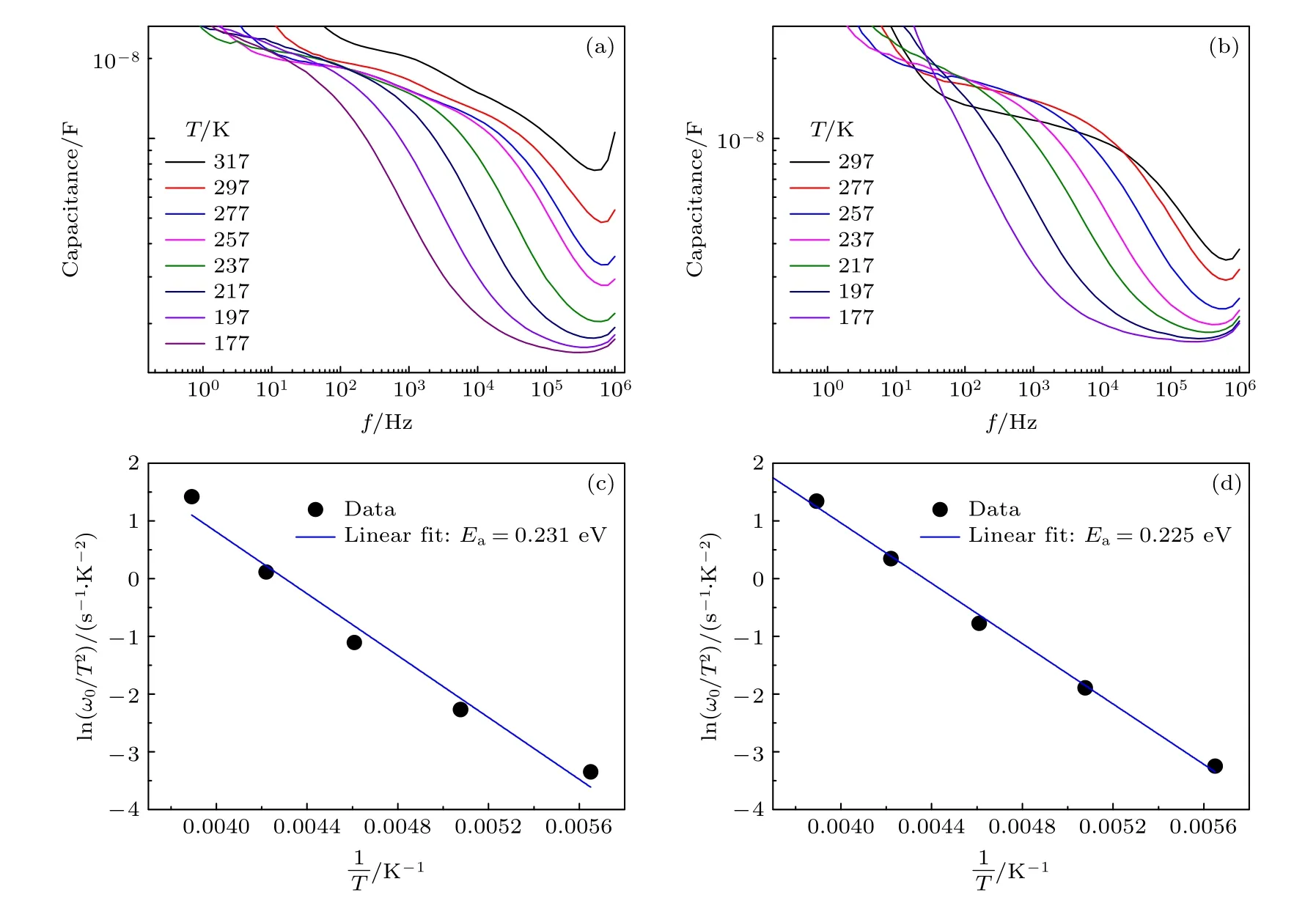
图10 不同温度下(a) CsBr 钝化后的电池和(b)无CsBr 钝化的电池的界面电容数值; (c) CsBr 钝化后的电池和(d)无CsBr 钝化的电池在特定测试频率下的Arrhenius 点, 基于此可获得缺陷的活化能[117]Fig.10.Temperature dependence of capacitance for (a) device with CsBr and (b) control device without CsBr.Arrhenius plot of the characteristic frequencies to extract the defect activation energy for (c) device with CsBr and (d) control device without CsBr[117].
其中ω0是逃逸频率.能量分界以下的陷阱态可以捕获或发射给定电荷并贡献电容, 根据系统的频率拟合结果得到CsBr 钝化后缺陷态浓度从5 ×1016cm–3降低到2 × 1016cm–3.
尽管TAS 是一种相对有效的缺陷数量研究方法, 但实际上以整个PSCs 为研究表征对象, 并不能剔除体相缺陷对最后结果的影响.事实上, 正电子对原子尺度的缺陷特别敏感, 易被点阵中的负电荷捕获从而发生湮没, 大量湮没事件的总和就是正电子湮没寿命谱(τ).正电子在材料完整晶格周期性势场作用下的运动行为, 与在含有缺陷的晶格周期性势场下运动的行为不同, 且材料中的缺陷类型不同, 正电子在材料中的湮没会存在多种正电子寿命(τ1,τ2, ···), 通常正电子的寿命谱可以反映正电子所在处的局域电子密度、缺陷的类型和缺陷的浓度; 而正电子湮没辐射能谱的多普勒展宽可提供正电子湮没处电子动量的信息.正电子湮没技术有可能是一种有效的界面缺陷浓度研究方法.
5 空穴传输层的光照不稳定
最早常用的空穴传输材料为溶解有锂盐的Spiro-OMeTAD, 而Spiro-OMeTAD 的有机本质使其高温稳定性并不好, 且光照下电池表面温度很可能超过70 ℃, Sanchez 等[119]对比研究了光照对Spiro-OMeTAD 和钙钛矿薄膜的影响, 在空气和惰性氛围下, 吸收光谱的测试结果显示Spiro-OMeTAD 出现很明显的光氧化现象, 且锂盐的加入加速了其光氧化行为, 若Spiro-OMeTAD 穿过钙钛矿薄膜的孔隙与TiO2电子传输层直接接触,那么其降解进程将更加快速.Jena 等[120]在温度为60—120 ℃且相对湿度30%—50%的环境区间内研究PSCs 的失效机制, 试图重新利用钙钛矿材料中的铅元素避免铅污染, 且与Sanchez 等[119]有相同的发现, 即PSCs 的失效主要是钙钛矿/Spiro-OMeTAD 的界面在高温下的失效引起, 相反钙钛矿本身的高温不稳定行为并非电池失效的主要诱因, Spiro-OMeTAD 失效的过程中会产生大量孔隙, 这使得Spiro-OMeTAD 从成分上产生了不可逆转的破坏, 因此解决光辐照下Spiro-OMeTADl的热不稳定性也是研究重点.Matteocci 等[121]电池进行了封装, 隔绝了水和氧, 仅研究光照产生的热对电池稳定性的影响, 发现长时间照射会对PSC 性能产生影响, 60 ℃时, 效率降低为0.1%/h,85 ℃时, 效率降低为0.21%/h, 且效率的降低是由Spiro-OMeTAD 的失效引起, 同时必须要提的是Spiro-OMeTAD 的价格昂贵, 因此, 出于提高PSCs 热稳定性以及降低成本的双重考虑, 碳纳米管/聚合物复合材料(SWNTs)取代有机空穴传输层成为一个很经济的选择, 其可有效减缓由热产生的有机空穴传输材料的降解, 且其对水进入的阻力显著增强[122], 除此之外, 全无机空穴传输材料也是一个很有意义的研究方向, 研究结果表明基于CuSCN 的PSCs 保持了约60%的初始值, 在相同的条件下比Spiro-OMeTAD 制备的PSC 表现出优异的热稳定性[123].虽然材料和器件的热稳定性得到改善, 但研究者发现使用CuSCN 的器件在高温下降解比使用Spiro-OMeTAD 的设备更快, 且界面降解机制发生在钙钛矿吸光层与CuSCN 之间的异质结[124].Liu 等[125]在提高电池稳定性的过程中发现钙钛矿/CuSCN 界面存在明显的失效问题, 他们猜测CH3NH3I 和CuSCN 之间发生反应生成了CH3NH3SCN 和CuI, 如方程(6)所示, 所以从有机分子的设计也需兼顾考虑效率和稳定性的问题.
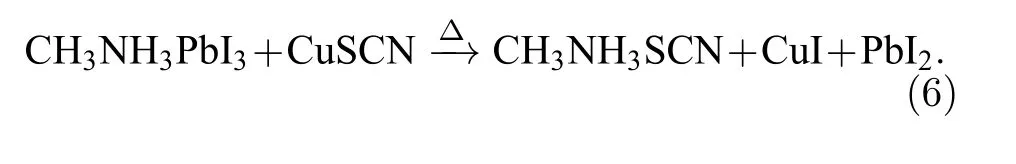
6 金属电极的光照不稳定
金属电极的光照不稳定主要体现在两方面, 一方面来自钙钛矿材料中的碘离子聚集在阴极界面会改变阴极的能级从而使电池性能下降, 且已在Ag 电极与钙钛矿材料的接触界面检测到碘化银,这说明金属电极可以与钙钛矿材料中的卤素离子发生反应; 另一方面Ag 和Au 等在光照条件下也会扩散到器件中, 在TiO2/SnO2界面附近积累导致电池失效, Li 等[126]使用ToF-SIMS 技术证实了金属元素在电池断面范围内的移动, 如图11 所示.金属电极的不稳定一方面可以产生分流跨越设备,减少器件的填充因子, 另一方面Ag 原子可以在钙钛矿薄膜内产生深陷阱态, 增强非辐射复合, 从而降解了Voc以及Jsc.Ming 等[127]使用第一性原理系统研究了Au, Ag, Cu, Cr, Mo, W, Co, Ni 和
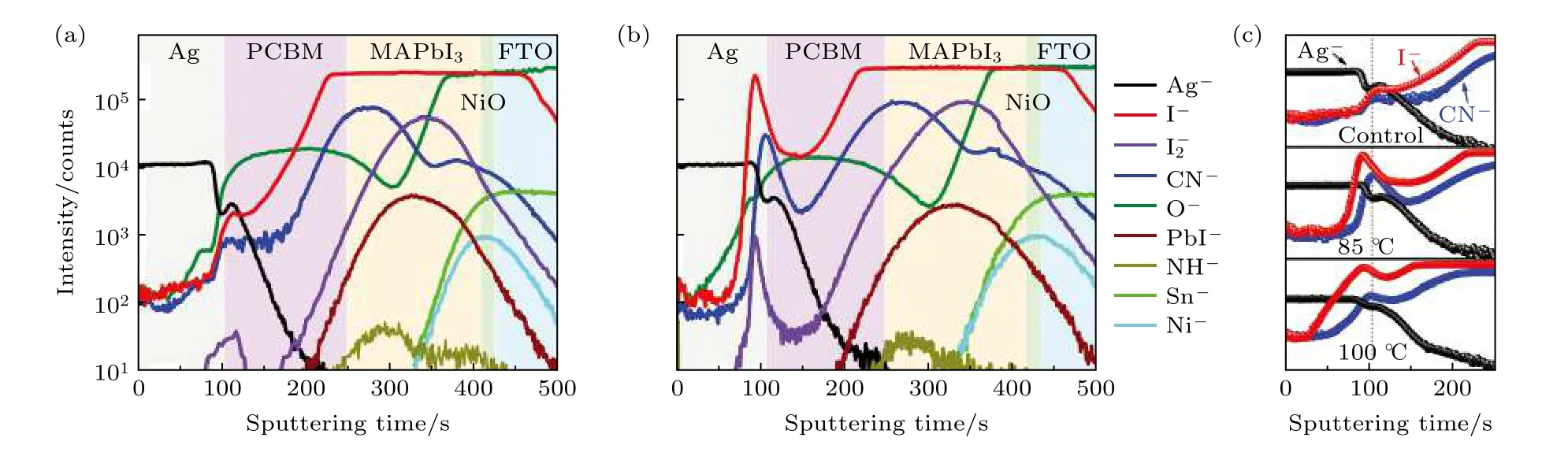
图11 在(a) 85 ℃下24 h 处理之前和(b)处理之后, 断面内ToF-SIMS 测试的元素分布; (c)热处理后不同温度下断面内Ag–,I–和CN 分布[126]Fig.11.ToF-SIMS elemental depth profiles (a) before and (b) after a thermal treatment at 85 ℃ for 24 h; (c) depth profiles of Ag–,I– and CN– after different temperature of thermal treatment[126].
Pd 作为金属电极, 当其原子插入到MAPbI3的晶格结构中后钙钛矿材料的磁性、形成能和扩散能垒的变化, 计算结果显示Mo 和W 电极因为较大的形成能和较高的扩散能垒而比Au, Ag 和Cu 电极稳定, Cr 电极的形成能虽然低但其扩散能垒很高,所以Cr 离子的迁移在动力学上也被阻止.但事实上, 一个优质的金属电极还要考虑其薄膜形态和逸出功与钙钛矿材料之间的匹配, 所以有很大一部分研究工作集中在空穴传输层与金属电极中加入离子迁移阻挡层或者碳电极的开发.
为了抑制钙钛矿与金属电极之间的离子传递造成的电池性能损失, Arora 等[128]将空穴传输材料Spiro-OMeTAD 替换为对离子传输更有阻挡力的CuSCN, 并在CuSCN 和Au 之间加入还原氧化石墨烯层, PSCs 在60 ℃的全太阳强度下, 老化1000 h 后, 仍具有95%的性能.同时, Arora等[128]在此基础上又发现用溶剂快速去除的方法,使CuSCN 层的微结构更加紧凑且致密, 以更高效的阻挡离子传递.Shao 等[129]则尝试将金属对电极替换成非金属材料, 用冷等静压法制备了2H-NbS2对电极薄膜, 2H-NbS2电导率高达8.7 × 103S/cm,功函数为5.20 eV.Yang 等[130]使用全碳柔性对电极来替代金属电极, 采用工业柔性石墨片和炭黑颗粒制作导电电极, 以保证电极与钙钛矿层接触良好, 最后以MAPbI3/TiO2为异质结太阳能电池获得了10.2%的效率.Zhang 等[131]在前者的基础上,同时替换空穴传输层和对电极, 他们使用酞菁铜(nanorod-liked copper phthalocyanine, CuPc)为空穴传输材料, 再加上可打印的低温碳作为对电极材料, 获得了16.1%的效率和更高的PSCs 稳定性.Liu 等[132]基于相同的思路, 使用p 型介观NiO作为空穴传输层和碳黑/石墨作为对电极制备了PSCs 效率为11.4%, 但相比较而言, 碳电极PSCs的电池效率仍然较低, 未来还有很多路要走.
7 总结与展望
近年来第一性原理的报道和电池光电性能测试的结果表明钙钛矿器件不能忽略离子迁移, 离子迁移造成的磁滞、荧光增强/淬灭以及电池失效都是PSCs 稳定性差的原因.不仅如此, TiO2/钙钛矿界面的紫外不稳定、空穴传输材料在光辐照下的热稳定性差及金属电极的光不稳定, 也是制约高性能PSCs 器件稳定性提高的关键因素.这些光辐照不稳定因素对于PSCs 稳定性提升有“木桶效应”,所以考虑到光生载流子作为光转换后的能量载体,有必要以光生载流子为研究对象, 描绘其从出现在钙钛矿薄膜内部到传输出外电路的全生命周期的传输复合行为, 归纳其在电池各个位置的影响因素, 比如在钙钛矿电池内部应耦合单纯的离子迁移以及离子迁移对电子传输的影响, 探索影响电子传输复合行为的各因素之间的相互影响制约关系, 明确主要影响因素以针对性的获得有效解决方案.这将为系统提高PSCs 光辐照稳定性提供理论依据,为进一步开发高效能结构和器件以及寻找新型光转换材料提供有力指导.
