先天性长QT综合征的分子基础和临床管理
2021-05-09王元丽吕铁伟
王元丽,吕铁伟
(重庆医科大学附属儿童医院心内科,重庆 400014)
提要:先天性长QT综合征(congenital long QT syndrome,cLQTS)是一种遗传性离子通道病,以QT间期延长为特征,患者易发生尖端扭转型室性心动过速而具有潜在致命风险,常表现为心悸、晕厥、心脏骤停、猝死。目前共发现16种cLQTS致病基因,LQTS1、LQTS2和LQTS3约占75%~85%,突变基因分别为KCNQ1(30%~35%)、KCNH2(25%~30%)和SCN5A(5%~10%),主要涉及缓慢延迟整流钾电流、快速延迟整流钾电流和钠电流,通过影响动作电位时程而增加心律失常易感性。明确诊断后根据危险分层选择合适的治疗方式,主要包括生活方式管理、药物治疗、左心交感神经切除术及植入型心律转复除颤器,基于分子机制的基因特异性治疗和个体化临床管理使cLQTS的治疗越来越精准有效。
先天性长QT综合征(congenital long QT syndrome,cLQTS)是一种以QT间期延长为特征的遗传性离子通道病,患者以心悸、晕厥、心脏骤停、猝死为主要临床表现,多于儿童及青春期发病,患病率约为1/2 000。cLQTS主要分为不伴有耳聋呈常染色体显性遗传的Romano-Ward综合征和常伴有神经性耳聋呈常染色体隐形遗传的Jervell-Lange-Nielsen综合征。目前共发现16种(见表1)致病突变,LQTS1、LQTS2和LQTS3约占75%~85%,突变基因分别为KCNQ1(30%~35%)、KCNH2(25%~30%)和SCN5A(5%~10%),中国最常见的类型是LQTS2[1]。明确cLQTS的突变基因及分子机制有助于疾病的诊断、分层及治疗,现就近年来cLQTS的分子机制和临床管理进行详述。
1 分子基础
1.1 缓慢延迟整流钾电流异常
LQTS1、LQTS5、LQTS11和Jervell-Lange-Nielsen综合征均与缓慢延迟整流钾电流(IKs)有关。IKs通道是外向电压依赖性门控钾通道,交感神经通过G蛋白偶联受体(G protein-coupled receptors,GPCRs)-腺苷酸环化酶(ade⁃nylylcyclase,AC)-环磷酸腺苷(cyclic adenosine monophos⁃phate,cAMP)-蛋白激酶A(protein kinase A,PKA)-KCNQ1信号转导通路以及A激酶锚定蛋白9(A-kinase anchoring protein,AKAP9)参与的信号转导通路使IKs功能上调,增加外向电流以抵消增加的内向钙电流,防止心肌动作电位时程延长以保证足够的舒张期[2]。IKs通道功能障碍使平台期复极化电流减少,心肌动作电位时程延长,心肌细胞易发生早期后除极而引发致命性心律失常,如尖端扭转型室性心动过速。
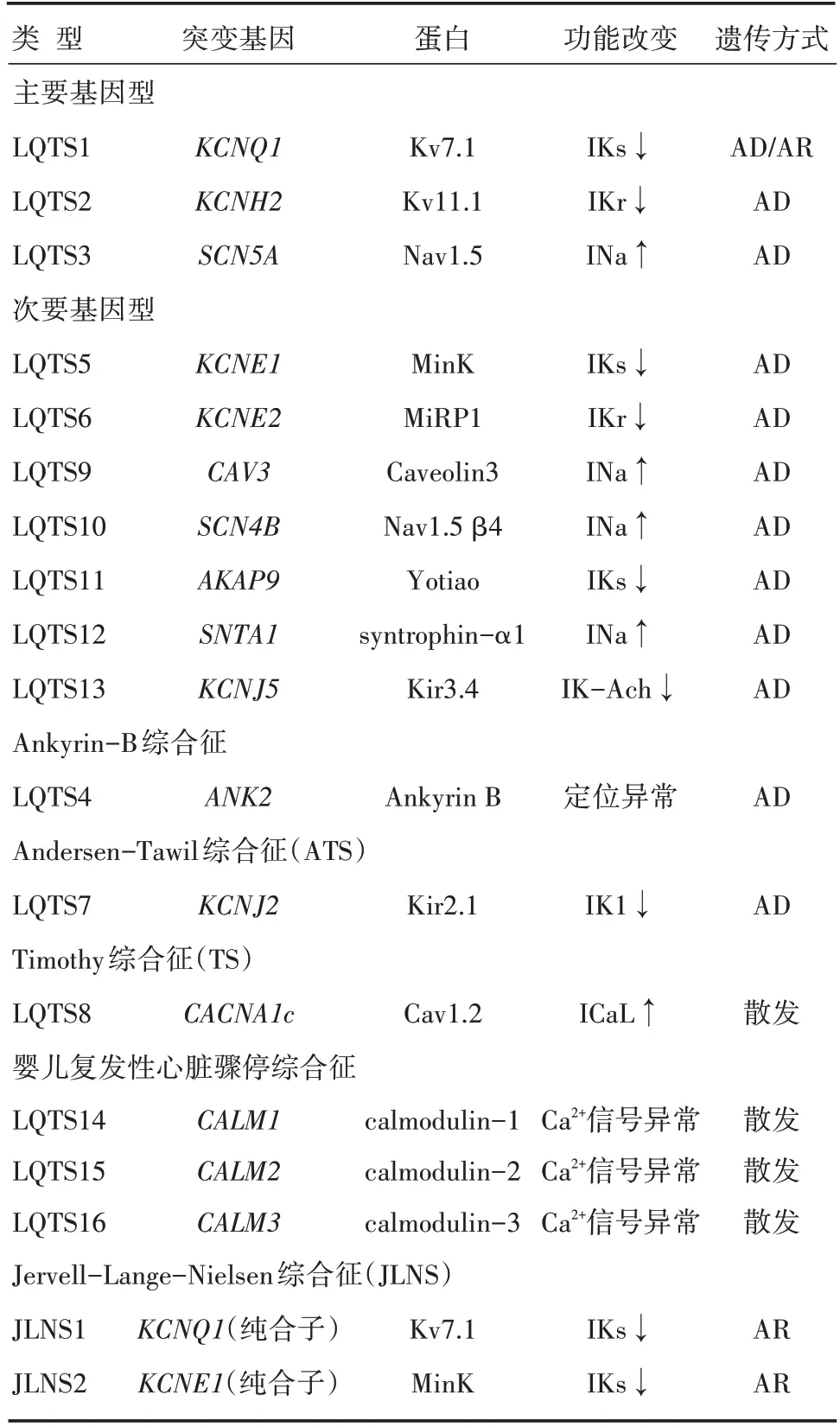
表1 cLQTS基因型
LQTS1的突变基因为KCNQ1,其合成Kv7.1蛋白组成IKs通道的α 亚基[3],通过各种分子机制导致IKs通道功能障碍。(1)离子渗透缺陷:突变主要发生在孔道结构域,选择性过滤器的构象发生改变,使K+渗透性降低,导致IKs电流幅度降低。(2)门控缺陷:孔道结构域突变、羧基端突变或电压感受域突变通过影响电压感受域和孔道结构域耦合、改变IKs电流-电压关系等方式影响通道门控特性,使IKs开放时间缩短[2]。(3)转运障碍:某些羧基端突变可减少通道蛋白向质膜表面的转运,某些氨基端突变会增加内质网潴留而减少通道蛋白在细胞膜表面的表达[4]。(4)PKA介导的信号转导异常:某些突变会影响GPCRs-AC-cAMP-PKA-KCNQ1信号通路,使肾上腺素能刺激无法上调IKs功能[5]。(5)磷脂酰肌醇二磷酸(phosphatidylino⁃sitol 4,5-bisphosphate,PIP2)亲和性降低:PIP2结合在细胞质环和羧基端,形成电压感受域和孔道结构域之间的界面,稳定IKs通道开放状态。某些突变影响PIP2结合位点导致PIP2亲和性降低[2]。(6)KCNQ1-KCNE1相互作用阻断:某些羧基端突变破坏其与KCNE1羧基端的相互作用,可同时降低PIP2亲和性,导致电流幅度降低和电流-电压关系改变[6]。(7)钙调蛋白亲和性降低:钙调蛋白在离子通道蛋白的组装和转运中起重要作用,某些突变可降低钙调蛋白亲和性,使离子通道蛋白在细胞膜表面的表达降低[7]。大多数错义突变通过以上机制使IKs功能障碍,极少数无义突变或移码突变提前生成终止密码子导致IKs功能丧失。
KCNE1合成minK蛋白组成IKs通道的β 亚基[3],KCNE1基因突变可通过门控缺陷、转运障碍和KCNQ1-KCNE1相互作用阻断等机制影响IKs通道功能,从而导致LQTS5[6]。AKAP9蛋白(yotiao蛋白)结合在KCNQ1的羧基端,募集PKA、蛋白磷酸酶1、腺苷酸环化酶9和磷酸二酯酶PDE4D3等信号蛋白形成IKs大分子复合物,响应肾上腺素能刺激而严格控制通道磷酸化,使通道活化增快和失活减慢。AKAP9基因突变影响PKA与KCNQ1的相互作用,导致IKs上调障碍是LQTS11的分子基础[8-9]。
1.2 快速延迟整流钾电流异常
LQTS2突变基因是KCNH2,编码hERG蛋白组成快速延迟整流钾电流(IKr)的α 亚基,LQTS6突变基因是KCNE2,编码KCNE2蛋白(MiRP1蛋白)组成IKr的β 亚基。IKr通道也是外向电压依赖性门控钾通道,心肌细胞复极时激活,复极本身推动更多IKr通道活化,当细胞持续复极到静息电位时IKr通道缓慢钝化。静息电位时仍有一部分IKr通道保持开放状态,抑制细胞去极化,防止期前收缩和快速性心律失常,因此,IKr功能障碍易导致期前收缩性心律失常[2]。
hERG突变的致病机制包括以下4类。(1)hERG蛋白合成减少:无义介导mRNA衰变是一种细胞损伤控制机制,可破坏含有无义突变的mRNA,阻止翻译出缩短的hERG肽链,单倍剂量不足导致细胞无法维持正常的生理功能[10]。(2)转运障碍:某些突变导致hERG蛋白错误折叠而滞留在内质网,转运到质膜的hERG蛋白减少。hERG转运障碍见于大多数KCNH2错义突变,是IKr功能障碍最主要的机制[11]。(3)门控缺陷。(4)K+选择性或渗透性异常。
1.3 钠电流异常
钠电流(INa)主要由内向电压依赖性门控钠通道(Nav1.5)介导,激活后允许Na+快速进入细胞内去极化,后钠通道快速失活,心肌细胞进入复极化。钠通道失活具有高度电压依赖性,是造成心肌细胞不应期的原因,LQTS3、LQTS9、LQTS10和LQTS12均与Nav1.5无法正常失活有关。心室动作电位的平台期或复极期Nav1.5未能失活或未能维持失活状态,存在持续晚期电流(INaL)导致心肌动作电位时程延长。SCN5A基因编码Nav1.5的α亚基,其突变可导致少数钠通道不完全失活或失活的钠通道再开放从而引起LQTS3[12,13]。SCN4B编码Nav1.5通道的β 亚基,对Nav1.5失活具有重要影响,其突变可导致LQTS10[14]。CAV3基因和SNTA1基因分别编码caveolin-3和syntrophin-α1,二者均为钠通道辅助蛋白,其突变可促进钠通道复活、增加钠电流峰值从而产生持续INaL[2,15]。
1.4 其 他
锚蛋白B作为衔接蛋白确保离子通道及转运蛋白的稳定,对Na+/Ca+交换体、Na+-K+-三磷酸腺苷(adenosine triphosphate,ATP)酶的正确定位起重要作用,可调节电压门控钠通道和ATP门控钾通道。编码锚蛋白B的ANK2基因突变破坏正常的靶向功能,引起离子通道调节障碍,导致常染色体显性遗传病LQTS4[2,16]。KCNJ2基因编码内向整流钾通道Kir2.1,介导IK1电流,维持心肌细胞的静息电位,参与动作电位的末期复极化。PIP2与Kir2.1结合促进通道活化,KCNJ2基因突变可降低PIP2亲和性使通道活化障碍而导致Andersen-Tawil综合征[17-18]。
CACNA1C基因突变引起Timothy综合征,该基因编码电压门控钙通道的α1C亚基Cav1.2。该通道产生L型钙电流,CACNA1C突变显著破坏Cav1.2的电压依赖性失活,导致钙离子内流增加而延长心肌动作电位时程[19]。KC⁃NJ5基因编码G蛋白偶联的内向整流钾通道Kir3.4,Kir3.4和Kir3.1形成同源或异源四聚体产生乙酰胆碱依赖性钾电流(IK-Ach)。KCNJ5基因突变会减少Kir3.4和Kir3.1向质膜转运并降低电流幅度,使心律失常易感性增加而引起LQTS13[2,20]。
钙调蛋白介导细胞钙信号传导,影响L型钙通道的Ca2+依赖性失活和Nav1.5通道的快速失活,对KCNQ1的运输和组装也起重要作用。CALM1-3分别编码CALM1、CALM2和CALM3,其新发突变与严重QT间期延长、室性心律失常以及房室传导阻滞相关。这些突变位于钙调蛋白直接结合Ca2+的EF基序,使钙亲和力降低,导致钠电流、钾电流以及心脏钙动力学异常[21]。
2 临床管理
2.1 危险分层
危险分层用以判断cLQTS患者40岁前发生心脏事件(晕厥、尖端扭转性室性心动过速、心脏骤停或猝死)的风险,常用的是表现型与基因型相结合的危险分层(如表2)[22-23]。不同年龄段发生心脏事件的风险不同,12~14岁是重要的过渡时期。青春期前,男性LQTS1患者心脏事件发生风险较高且发病较早;青春期后,女性LQTS2患者的风险增加,尤其是孕期或产后[24]。发生心脏事件的cLQTS患者Tpeak-Tend间隔明显延长[25]。多基因突变、大片段基因重排常导致严重的临床表现。突变位置在细胞质环的KCNQ1患者发生心脏事件的风险增加,通常由运动引发,但β 受体阻断药能显著降低风险[26]。KCNH2孔道结构域突变、CALM1或CALM2突变均会增加心脏事件发生的风险[27-28]。

表2 cLQTS危险分层
2.2 生活方式管理
LQTS1患者应避免剧烈活动,特别是游泳或跳水,LQTS2患者应避免暴露在突然的声音刺激(闹钟声、手机铃声等)下,所有患者应尽量避免严重电解质紊乱,避免服用延长QT间期的药物,如Ⅰ类和Ⅲ类抗心律失常药、喹诺酮类抗生素和三环类抗抑郁药等。最新研究证明致心律失常药物在cLQTS患者中的使用非常普遍并随着诊断时间延长而增加,提示临床医生应该提高对致心律失常药物的认识,重视对cLQTS患者的健康教育[29]。
2.3 药物治疗
β 受体阻断药是cLQTS患者的首选药物,能显著降低心脏事件发生率。对LQTS1效果最好,能够有效降低95%的心脏事件;对LQTS3疗效最差,其对LQTS3患者是否具有保护性作用仍有争议。不同类型的β 受体阻断药疗效不同,纳多洛尔较普萘洛尔、阿替洛尔强,美托洛尔效果最差,因此,美托洛尔不推荐用于cLQTS的治疗;纳多洛尔是唯一一个能够显著降低LQTS2心脏事件发生率的β 受体阻断药[30-31]。
近几年主张使用钠通道阻滞剂做为LQTS3的基因特异性治疗,氟卡尼、雷诺嗪和美西律可分别缩短静息QTc 10%、11%、12%。氟卡尼和雷诺嗪均可降低峰值INa和INaL,但在心室肌中雷诺嗪可选择性地阻断INaL,因此,发生Brugada综合征的可能性较氟卡尼低。现有数据证明氟卡尼和雷诺嗪主要适用于SCN5A-D1790G突变,美西律无突变位点依赖性[32-33]。
2.4 侵入性治疗
左心交感神经切除术通过切除左侧星状神经节可明显提高心室颤动阈值,有效降低高风险患者心脏事件发生的可能性。目前采用胸腔镜下左心交感神经切除术,减少了心室水平去甲肾上腺素的分泌,损伤小,术后常见并发症(霍纳综合征)的发生率降低,有效性和安全性都得到了保障[34]。近几年有应用经皮肾去交感神经导管射频消融术(renal sympathetic denervation,RSD)治疗cLQTS的病例报道,RSD通过高选择性去除肾动脉周围部分传入和传出神经,通过下丘脑中枢神经反馈机制,减少肾脏局部和全身去甲肾上腺素溢出,与左心交感神经切除术一样,降低心脏交感神经活性达到预防室性心律失常的目的[35]。植入型心律转复除颤器(implantable cardioverter de⁃fibrillator,ICD)可有效防止心脏骤停、猝死等恶性心脏事件,但对于无症状患者,首选β 受体阻断药治疗而不常规置入ICD。
3 结语
由于涉及离子通道种类多、信号转导分子和调节因子复杂,cLQTS的分子机制尚需进一步研究。诱导性多能干细胞技术和高通量膜片钳技术的发展,为我们研究致病基因的分子机制、基因型-表现型关系、基因修饰因子的作用以及基因特异性治疗提供了新的技术平台。cLQTS患者需要个体化的临床管理,临床医生需结合致病基因及临床信息进行全面有效的危险分层,鼓励患者积极改变生活习惯,优先药物治疗并规律随访,严格把握侵入性治疗的指针,避免过度ICD植入。
