熔融制样–X-射线荧光光谱法测定伟晶岩中主次量组分
2021-05-07赵亚男王小强杜天军余文丽杨惠玲
赵亚男,王小强,杜天军,余文丽,杨惠玲
(1.河南省有色金属地质勘查总院,河南省有色金属深部找矿勘查技术研究重点实验室,郑州 450052;2.河南有色金属地质矿产局第七地质大队,郑州 450018)
岩矿分析是对矿物岩石的成分在不同赋存形态下的含量及其化学组成进行分析测定[1]。伟晶岩是一种稀有金属矿床的重要赋矿主岩[2],由硅酸盐残浆侵入到岩浆岩或围岩裂隙中缓慢结晶而成,其本质为铝硅酸盐岩石,组成复杂,种类繁多[3],其中SiO2、Al2O3、Fe2O3、MgO、TiO2、K2O、Na2O、CaO 的总含量占90%以上[4],硅酸盐岩石的常规检测方法为化学法[5],以重量法为基础[6],还有容量法[7]、滴定法[8]、比色法[6]、原子吸收光谱(OES)法[9]等,此类方法操作繁琐,分析周期长,分析结果易受多种因素影响[10]。ICP–OES 法[11]虽然可以多元素同时分析,但只能测定少数几种低含量元素,而且前处理溶样过程比较繁琐[12]。
作为一种常规元素定性、定量分析方法[13],X-射线荧光光谱(XRF)法具有分析速度快、准确度和精密度高、自动化程度高、制样简单等特点[14],能同时测定多种元素,在地质样品分析中应用广泛[15]。XRF 光谱分析常用压片法和熔融法[16],XRF 分析硅酸盐岩石样品主次量组分已有报道[17–20],但伟晶岩样品XRF 分析检测鲜见报道。伟晶岩具有SiO2、Al2O3、K2O、Na2O 含量高,MnO、TiO2、Fe2O3、CaO、MgO 含量低的特点,其高含量K2O、Na2O 在实际检测时会引起干扰和偏离。
笔者采用熔融玻璃片法制样,利用一系列试验选择合适的测试条件,提高方法的精密度和准确度,消除样品粒度效应、基体增强吸收效应与共存元素的干扰,可以同时测定硅、铝、铁、钛、钾、钠等10 种主次量元素,取得了满意的结果。
1 实验部分
1.1 主要仪器与试剂
X-射线荧光光谱仪:ZSX Primus Ⅱ型,4.0 kW端窗铑靶X-射线管,真空光路,视野光栏Ф30 mm,超薄铍窗(30μm),衰减器为1/1,日本理学公司。
X 荧光光谱分析专用全自动熔样机:CNRY–02C 型,洛阳特耐实验设备有限公司。
电子分析天平:AL104 型,感量为0.1 mg,梅特勒托利多仪器(上海)有限公司。
铂– 黄坩埚:材质为铂金合金(质量比为95∶5),规格为30 mL,高25 mm,下端内径为32 mm,上端内径为45 mm。
四硼酸锂–氟化锂混合溶剂:优级纯,质量比为13∶1,X-射线荧光光谱分析专用,于700 ℃灼烧2 h,冷却后转入试剂瓶中,置于干燥器内备用,洛阳耐火材料研究院有限公司。
氯化钾:基准试剂,天津化学试剂厂。
硝酸铵:分析纯,洛阳化学试剂厂。
溴化铵:分析纯,天津市科密欧化学试剂开发中心。
火成岩标准物质:(1)编号分别为GSR–1、GSR–2、GSR–3、GSR–4、GSR–5、GSR–6,地球物理地球化学勘查研究所;(2)编号分别为GBW 07123、GBW 07125,国家地质实验测试中心;(3)编号分别为GBW 03116、GBW 03134,国家建筑材料研究中心。以上标准物质各组分质量分数见表1。
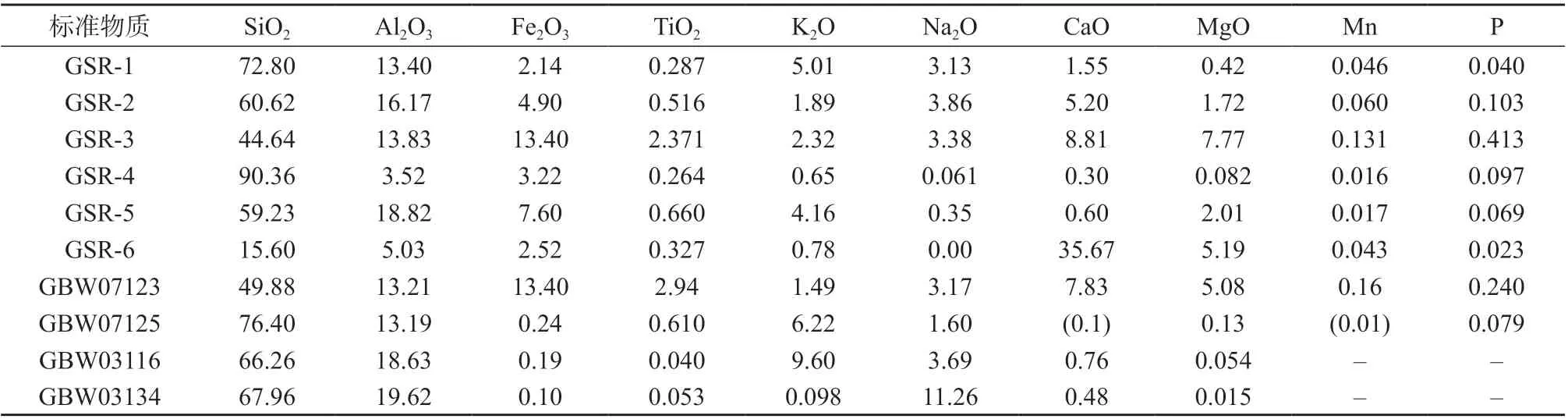
表1 火成岩标准物质各组分质量分数 %
钠钙硅玻璃标准物质:编号为GBW 03117,其中各组分质量分数见表2,国家建筑材料研究中心。

表2 钠钙硅玻璃标准物质各组分质量分数 %
土壤成分分析标准物质:编号分别为GSS–1、GSS–3、GSS–5、GSS–6、GSS–7、GSS–8,以上标准物质中各组分质量分数见表3。地球物理地球化学勘查研究所。
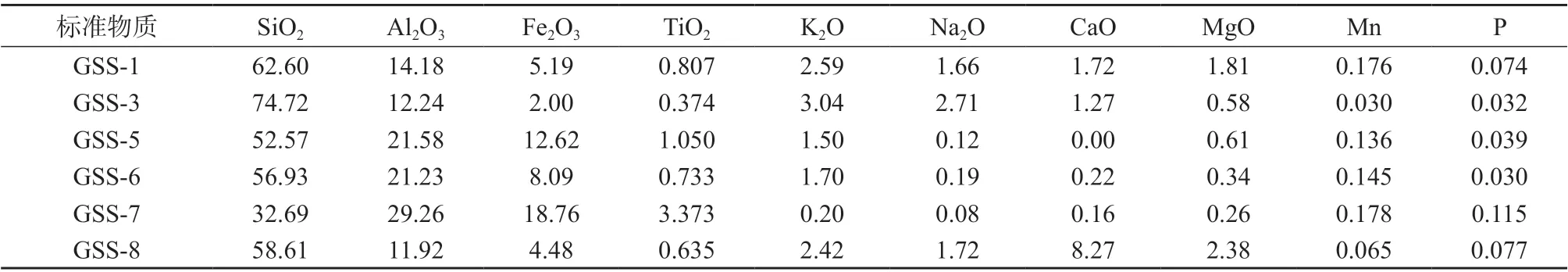
表3 土壤成分分析标准物质各组分质量分数 %
1.2 仪器工作条件
谱线:Kα;电压:50 kV;电流:60 mA。各组分测量条件见表4。
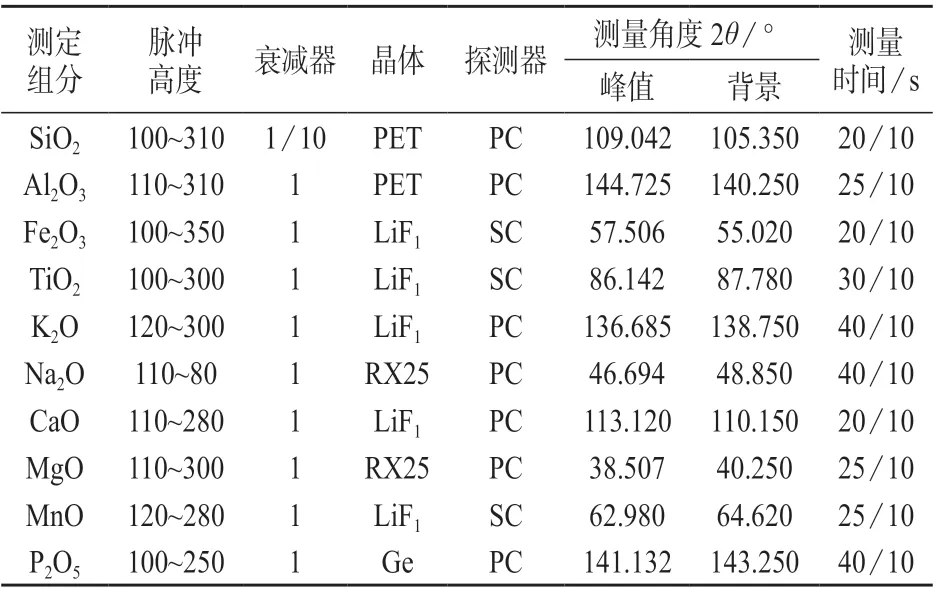
表4 伟晶岩各组分的分析条件
1.3 样品制备
将样品预先粉碎至74 μm(过200 目筛),在105 ℃烘箱内干燥4 h,取出,放入干燥器内冷却至室温,在分析天平上称取(8.000 0±0.000 5)g 四硼酸锂–氟化锂混合溶剂与(0.800 0±0.000 2) g 已烘干样品于铂–黄坩埚内,用细木棒搅匀,加入2 mL氧化剂(500 g/L 硝酸铵溶液),用胶头滴管滴加10滴脱模剂(饱和溴化铵溶液)。将铂–黄坩埚置于高频熔样机内,于700 ℃预氧化3 min,于1 200℃熔融4 min,熔融过程中自旋摇动,使坩埚内熔融物自动混合均匀,熔样结束后,随即用坩埚钳取出坩埚,此时观察坩埚内熔融样品底部是否有气泡,若有气泡可均匀转动摇晃将气泡赶出,静置,自然冷却。降温过程中熔融样片会发出“啪啪”响声,与坩埚自动脱离,成为直径32 mm 的透明圆片,如图1,背面贴上标签,放入干燥器内部保存待测。

图1 熔融制备的透明圆片样品
1.4 标准样片制备
选用火成岩标准物质、钠钙硅玻璃标准物质、土壤成分分析标准物质作为标准样品,制样方法同1.3。
混合标准样品1:采用钾长石标准物质GBW 03116 与基准试剂KCl 混合的方法配制4 个混合校准样品,其中K2O 质量分数参考值分别为19.2%、16.8%、14.4%、12.0%。
混合标准样品2:采用钠长石标准物质GBW 03134 与基准试剂KCl 混合的方法配制5 个混合校准样品,其中Na2O 质量分数参考值分别为10.13%、9.01%、7.88%、6.76%、5.63%。
1.5 样品测定
将待测样片装入样品盒中,置于X-射线荧光光谱仪工作台设定位置上,在1.2 仪器工作条件下,测定各元素荧光强度,利用标准工作曲线法定量,经基体矫正因子校正后,仪器自动给出元素含量。
2 结果与讨论
2.1 制样方法的选择
X-射线荧光光谱法一般采用粉末压片和熔融制样两种方法。粉末压片尽管制样简便、快速、成本低廉,但容易受到试样颗粒度、矿物效应和结构效应等不均匀效应的影响,尤其对轻元素分析影响较大。熔融制样可以消除X-射线荧光分析中的颗粒度、矿物和结构效应等不均匀效应的影响,提高主、次量元素(尤其是轻元素)的测定精度和准确度。另外,通过熔融制样,采用高纯试剂按一定比例配制校准样品,可以解决某些种类试样缺少标准物质的困难,故采取熔融制样法制备样品。
2.2 熔剂及其稀释比例的确定
X-射线荧光光谱分析常用熔剂有四硼酸锂、偏硼酸锂、碳酸锂。单纯用熔剂熔片虽然简单快速,但因缺少氧化剂和助溶剂(氟化锂),用这种熔片方法熔融时颗粒分解不完全,会导致结晶、开裂或破碎,使熔片作废[21]。四硼酸锂–氟化锂熔点较低,熔样均匀,流动性和重现性好,故选择四硼酸锂–氟化锂混合熔剂。
实验结果发现,熔剂稀释比例对熔样效果影响较大。当稀释比小于1∶10 时,虽然有利于提高荧光强度,但熔融物流动性差并有气泡残留,熔片厚度不均匀,样品不能完全熔融,表面形成悬浮物或者熔片出现裂纹。当稀释比大于1∶10 时,样品在融通过程中流动性较好,并且静置冷却后容易自动脱离坩埚,熔片厚度均匀。但不宜继续扩大稀释比例,因为尽管这样能提高熔片质量,但低含量元素的检测灵敏度较低。因此选定样品与熔剂的稀释比例为1∶10,即样品取0.800 0 g,熔剂取8.00 0 g。
2.3 脱模剂与氧化剂及用量的确定
卤素是最有效的脱模剂元素,常用的脱模剂有溴化铵、碘化铵、溴化锂与碘化锂,在熔样之前一般将脱模剂溶液加入到混合物中,或在熔融期间加入固体盐。碘化物和溴化物多数可以使用溶液,碘化铵与碘化锂因不容易吸潮而需要使用固体,而溴化锂溶解性好,只能作为溶液使用[22]。溴化铵、碘化铵比溴化锂、碘化锂更容易挥发,碘化铵中的碘对钛存在谱线重叠干扰。经综合考虑,实验选择溴化铵作为脱模剂。
通常在熔融之前将脱模剂溶液1 滴或数滴加入到坩埚中待熔融混合物表面。脱模剂的用量直接影响熔片的质量,脱模剂的常规用量大约为熔剂质量的0.2%~0.4%,脱模剂用量少会导致流动性差、熔片不均匀;而脱模剂用量过大会导致熔片过分收缩,呈不规则形状。脱模剂的最佳用量取决于样品的性质、熔剂组成及样品与熔剂的混合比例。确定脱模剂最佳用量的方法是:如果熔融样片呈现均匀圆形透明状,表面光滑并容易自动脱离坩埚,说明加入量合适;如果熔融样片形状不规则,表面凹陷或者有气泡、裂纹出现,不容易自行脱离坩埚,则可通过控制脱模剂的加入量来避免。多次实验结果表明,熔融前在待熔融混合物表面均匀加入10 滴溴化铵饱和溶液,容易制得均匀透明圆形熔片。
在熔剂中添加氧化剂的作用是氧化样品中少量的有机质和还原性物质,从而减少坩埚的腐蚀。常用氧化剂有硝酸铵、硝酸钠、硝酸锂等,由于选择溴化铵作为脱模剂,所以选用硝酸铵作为氧化剂以避免其它基体干扰。实验选择加入2 mL 500 g/L硝酸铵溶液,避免过多的氧化剂造成浪费。
2.4 基体效应和谱线重叠校正
基体效应是XRF 分析中难以避免的问题,是元素分析的主要误差来源。由于地质样品各元素含量范围宽,虽经熔融制样消除粒度效应,但元素间依然存在相互影响,基体效应仍然存在。本法使用日本理学公司软件提供的校正曲线和基体校准一体回归方程计算各组分基体效应的校正系数和谱线重叠干扰校正系数[23]。
2.5 漂移校正
仪器漂移或者短期波动会引起光谱分析误差,分析前需进行漂移校正[17]。
2.6 线性方程、线性范围与方法检出限
按照实验方法对标准样品进行分析,以元素质量分数为横坐标,以荧光计数率为纵坐标,绘制标准工作曲线,其线性回归方程、线性范围、相关系数见表5。由5 可知,各元素标准工作曲线线性相关系数均大于0.999 5。
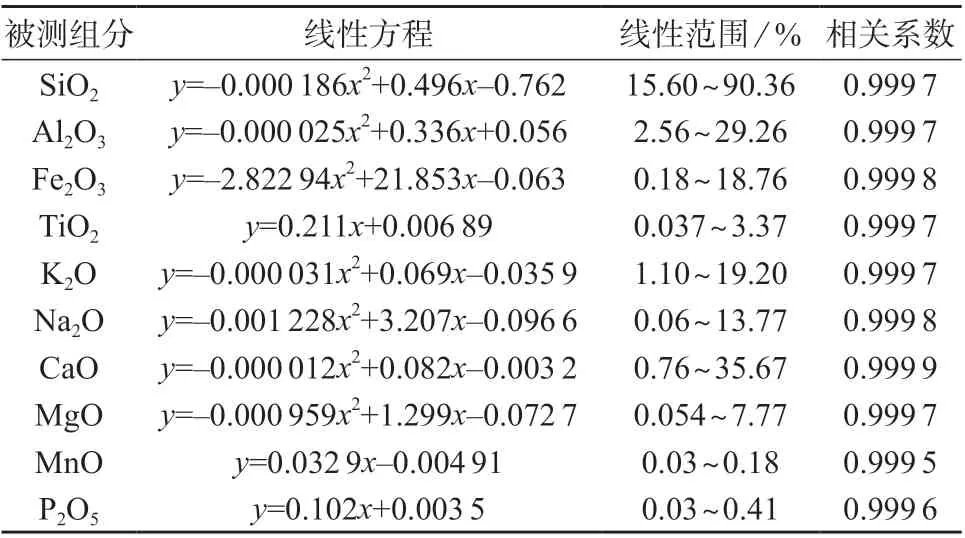
表5 各元素线性方程、线性范围、相关系数
检出限是分析测试的重要指标,与分析空白值、精密度和灵敏度密切相关,对于仪器性能的评价和方法的建立都是重要的基本参数之一。检出限与测量时间的选取及样品基体相关,测量时间越长,检出限越低;不同的样品因其组分和含量不同,散射的背景强度及分析元素的灵敏度都会发生变化,因而检出限也不同[14]。
根据分析元素的测量时间测定并计算方法检出限[17],结果列于表6。
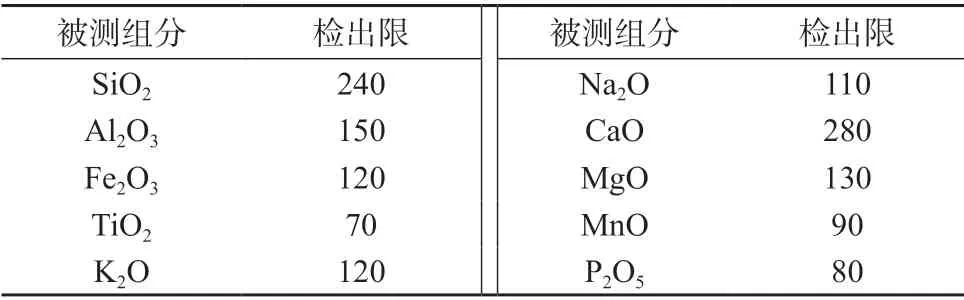
表6 各元素的检出限 μg/g
2.7 精密度试验
选用同一伟晶岩样品,按照实验方法进行12 次重复熔融测定,测定结果列于表7。
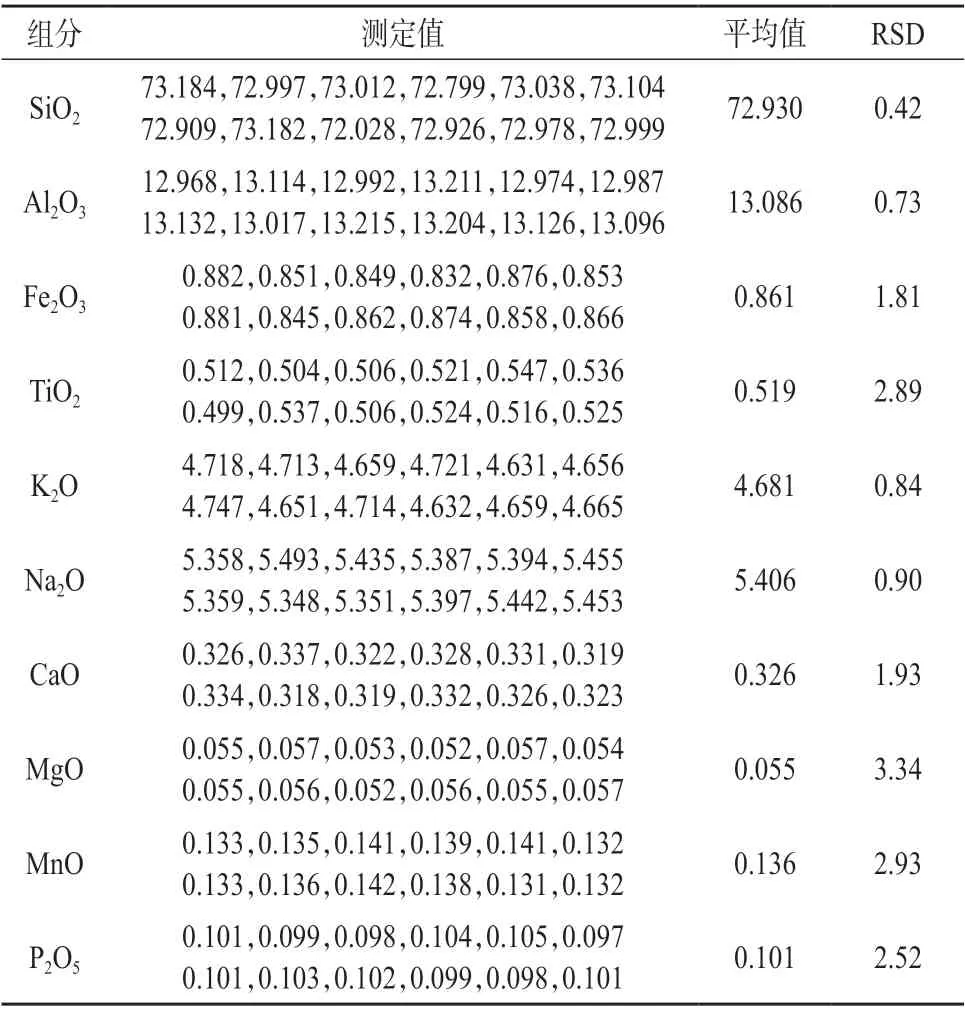
表7 精密度试验结果 %
由表7 可知,各组分测定值的相对标准偏差为0.42%~3.34%,表明本方法精密度较高。
2.8 准确度试验
分别对国家标准物质GBW 07123 和GBW 07125 进行重复测定,测定结果列于表8。由表8 可知,本法测定结果与标准值基本吻合,相对误差不超过10%,表明本法准确度较高。
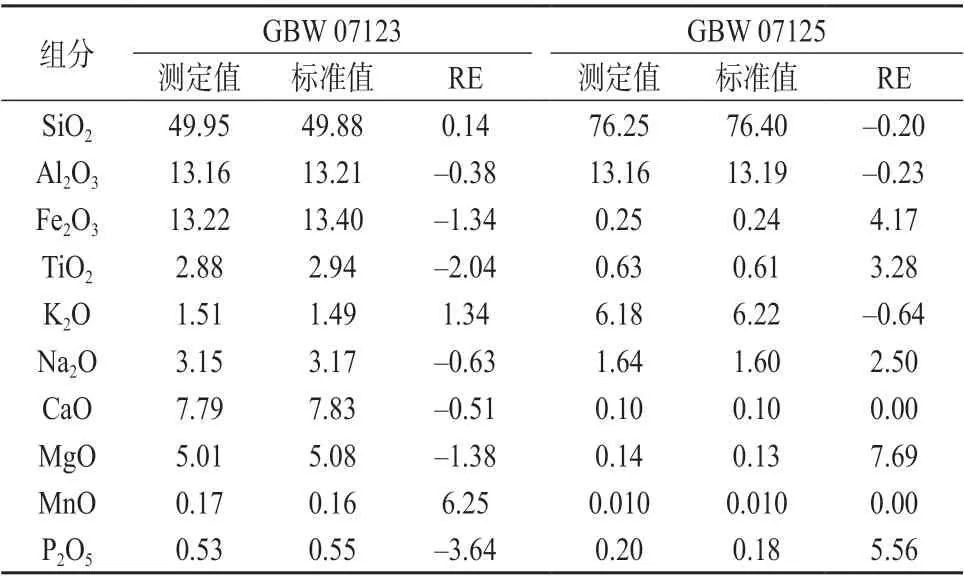
表8 准确度试验结果 %
3 结语
采用国家标准物质与自制混标模式结合的方法,拓宽待测元素测定范围,熔融制样,建立了XRF分析伟晶岩中主次量组分的分析方法。与传统的重量法、滴定法、分光光度法等方法相比,本法具有制样简单、适用范围宽、准确度与精密度高,可同时测定多种元素,简便快捷,满足快速检测的要求。
