溴代双Salamo型Zn(II)配合物的合成、结构及荧光性质
2021-04-28刘国华谢克锋李亚娟
刘国华,张 阳,谢克锋,李亚娟
(兰州交通大学 化学与生物工程学院,兰州 730070)
Salen型配体及其衍生物在现代配位化学中发挥着重要的作用[1],Salen型配体与不同的金属离子相互作用后可形成结构迥异,性质不同的金属配合物[2].因此,许多科学家对这些Salen型配合物的发光性能、催化性能、磁性质、电化学传导性和非线性光学材料等方面进行了深入的研究[3-13].但是Salen型化合物的稳定性较差,为了提高Salen型化合物的稳定性,近年来,报道了许多用-CH=N-O-(CH2)n-O-N=CH-单元替代Salen型双肟配体的-CH=N-(CH2)n-N=CH-单元从而形成Salamo型配体的研究[14-18],配体单元中的N2O2配位环境可与d区和f区金属原子配位后产生结构新颖、性质多变的配合物.基于此,Nabeshima课题组和我们课题组分别对Salamo型配体及其配合物进行了广泛的研究[19-21],发现此类配体及其配合物具有良好的发展潜力,有望成为配位化学新的研究热点之一.
双Salamo型配体与Zn(II)离子结合过程中常伴随化学键空间位置及荧光性质的变化,研究这一配位反应可为环境中Zn(II)离子的识别与定量检测提供基础理论依据.基于此,本文设计并合成了以2,3-二羟基-1,4-萘二醛为母体,3,5-二溴水杨醛修饰末端基的双Salamo型四肟配体(H4L),并以此配体与Zn(OAc)2·2H2O在乙醇和二氯甲烷的混合溶剂中反应,通过自然挥发法得到该配合物单晶体[Zn3L(AcO)2(EtOH)][Zn3L(AcO)2(H2O)2].通过元素分析、溶解性实验、红外光谱分析和X-射线单晶衍射等手段对配合物进行了表征,确定了其空间结构,并通过Hirshfeld表面分析和DFT理论计算对配合物进行了理论计算分析,通过荧光光谱分析了该配体与Zn(II)配合物的荧光性质.以上研究可为双Salamo型配体与Zn(II)离子配位的理论研究和实际检测识别提供可靠的实验依据.
1 实验部分
1.1 实验试剂与仪器
实验所用的化学试剂如表1所列.实验仪器:Sartorius CP225D 电子秤;恒温磁力搅拌器;旋转蒸发仪RE-2000B;SHZ-DIII循环水真空泵;三用紫外分析灯;北京泰克X-4显微熔点测定仪;德国Bruker VERTEX 70型红外光谱仪;X-射线单晶衍射仪等.

表1 试剂目录
1.2 配体H4L的合成
1,2-二胺氧乙烷合成方法见文献报道[22].相应的反应方程式如图1所示.
称取1,2-二胺氧乙烷0.184 g (2 mmol)溶于10 mL乙醇得溶液1,另称取3,5-二溴水杨醛0.56 g (2 mmol)溶于20 mL乙醇得溶液2,将溶液2置于恒压滴液漏斗中,然后缓慢滴加至溶液1中,反应温度55 ℃,加热搅拌6 h,冷却至室温后旋干溶剂得白色固体,并通过柱层析分离,得到单缩化合物3.将溶有2,3-二羟基-1,4-萘二醛的无水乙醇溶液置于恒压滴液漏斗中,然后缓慢滴加至溶有单缩化合物3(0.704 g,2 mmol)的无水乙醇溶液中,混合液在温度60 ℃下反应24 h,减压蒸馏至有白色固体析出,过滤,烘干得目标配体H4L.反应方程式如图2所示.

图1 1,2-二胺氧乙烷的合成Fig.1 Synthesis of 1,2-diaminoxyethane
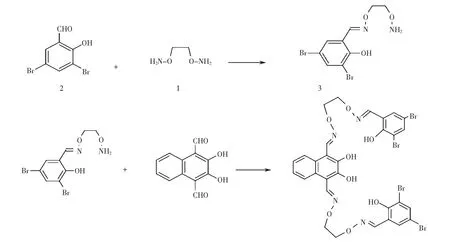
图2 配体H4L的合成路线Fig.2 Synthetic route to the ligand H4L
1.3 Zn(II)配合物的合成及单晶培养
Zn(II)配合物的单晶体采用溶剂自然挥发法得到.室温下,将Zn(OAc)2·2H2O (6.60 mg,0.03 mmol)溶于2 mL无水乙醇溶液中,另称取H4L配体(8.83 mg,0.01 mmol)溶于4 mL二氯甲烷溶液中,将上述两种溶液混合后颜色立即变成黄色.溶液磁力搅拌1 h,过滤,滤液封口静置,10天后,溶剂部分挥发后得到了橙黄色透明单晶体,分子式:C70H61Br8N8O27Zn6.
1.4 X-射线单晶衍射数据
Zn(II)配合物晶体结构用SHELXTL程序算得,运用Patterson函数法求得金属原子坐标,经Fourier合成进行多次迭代得到全部非氢原子坐标,再对所有非氢原子坐标和各向异性参数进行修正,其中氢原子由理论加氢得到.该配合物的单晶近似尺寸为0.23 mm×0.21 mm×0.17 mm,通过Bruker Smart 1000 CCD面探测衍射仪检测,X-射线衍射数据通过石墨单色Mo-Kα辐射在154.89(10)K温度下,用ω扫面方式采集.配合物CCDC:2055963,其键长、键角参数如表2所列.
2 结果与讨论
2.1 元素分析
Zn(II)配合物的元素分析数据及组成见表3.由表3可以看出,该配合物的所有元素分析结果与理论值很接近.

表2 Zn(II)配合物的晶体学数据
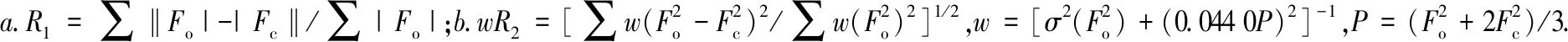
2.2 溶解性测定
室温下,测定了Zn(II)配合物在9种有机溶剂中的溶解性情况,结果见表4.由此可得:配体H4L与乙酸锌(II)形成的Zn(II)配合物在9种有机溶剂中的溶解性差异较大,易溶于DMF和DMSO等强极性溶剂,在非极性溶剂中溶解性较差.
2.3 红外光谱分析
配体H4L及其Zn(II)配合物的红外光谱在500~4 000 cm-1区域内显示出不同的峰,详见表5.
自由配体H4L中的νC=N键的伸缩振动吸收峰出现在1 610 cm-1处,而在Zn(II)配合物中,νC=N键的伸缩振动吸收峰向低波数位移,出现在 1 602 cm-1处.相对于自由配体,这一吸收峰向低波数位移了8 cm-1,说明该配体有较强的π-接受能力,也表明配体的C=N基团中的氮原子与Zn(II)离子发生了配位.配体H4L在3 421 cm-1处的吸收峰为酚羟基O-H伸缩振动吸收峰,而在Zn(II)配合物中该位置的吸收峰消失了,说明配体H4L中的酚羟基去质子后与Zn(II)离子配位了.此外,在配合物中观察到3 357 cm-1和3 409 cm-1处出现的吸收峰可分别归属为参与配位的乙醇分子和水分子的O-H伸缩振动谱带.
在配体H4L中,νAr-O的伸缩振动吸收峰位于1 258 cm-1处,在配合物中此吸收峰出现在1 226 cm-1处.νAr-O的伸缩振动吸收峰向低波数发生了位移,可归因于Zn(II)离子和酚氧原子之间形成了Zn-O键.此外,配合物在530 cm-1处出现了M-O键的吸收峰,而在自由配体H4L中未出现,也表明了配体与乙酸锌发生了配位.
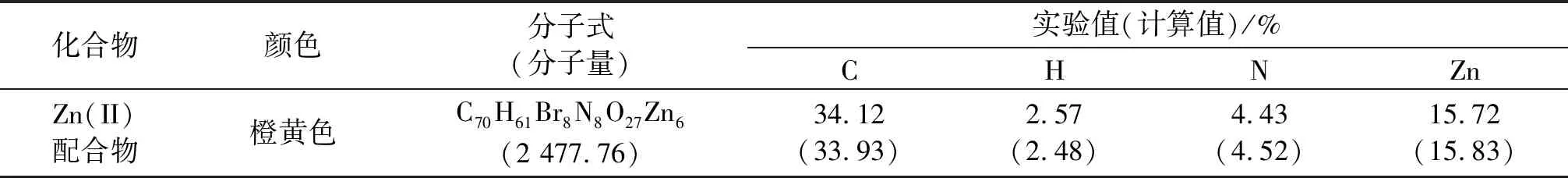
表3 Zn(II)配合物的元素分析数据

表4 Zn(II)配合物在有机溶剂中的溶解性测定
2.4 Zn(II)配合物的晶体结构分析
Zn(II)配合物的晶体结构及金属原子的配位多面体如图3所示.X-射线单晶衍射结构数据表明,Zn(II)配合物晶体的空间群为P21/n,属于单斜晶系.该晶体[Zn3L(AcO)2(EtOH)][Zn3L(AcO)2(H2O)2]由两个化学不等同,晶体学不等同的独立三核结构单元(A和B)组成,其中,A部分由一个完全去质子化的配体(L)4-单元、三个Zn(II)原子、两个桥联的醋酸根离子,以及参与配位的一个乙醇分子构成;B部分由一个完全去质子化的配体(L)4-单元、三个Zn(II)原子、两个桥联的醋酸根离子,以及参与配位的两个水分子构成.
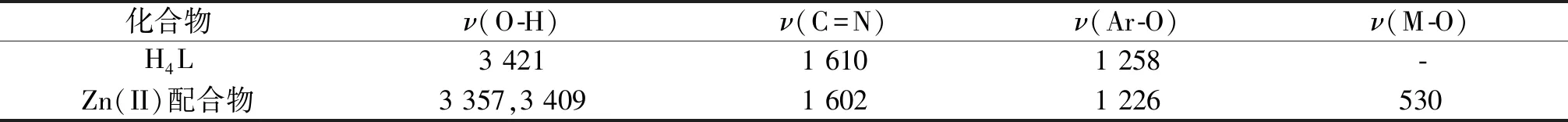
表5 配体H4L和Zn(II)配合物主要的红外光谱数据
如图3(a)所示,A部分两端的两个Zn(II)原子分别与配体提供的两个肟氮原子、两个酚氧原子以及双齿桥联醋酸根中的一个氧原子配位,而处于中心的Zn(II)原子分别与配体的两个酚氧原子、一个乙醇分子的氧原子和双齿桥联醋酸根中的两个氧原子配位.Zn1原子是五配位的,与配体中两个完全去质子的酚氧原子(O1,O4)和肟氮上的两个氮原子(N1,N2)以及桥联醋酸根上的一个氧原子(O12)配位.与之对称的Zn3原子也是五配位的,配位模式同Zn1一样.中心Zn2原子与配体中两个完全去质子的酚氧(O4,O5)、一个乙醇分子的氧原子(O13)和双齿桥联醋酸根上的两个氧原子(O10,O11)配位,形成五配位构型.配合物中配位金属原子的空间构型可通过计算τ值来确定,公式:τ=(最大键角-第二大键角)/60,表6给出了Zn(II)配合物的主要键长与键角.依此计算可知,三个Zn(II)原子的空间构型均为三角双锥(见图3(b)).
同样的,如图3(a),B部分两端的两个Zn(II)原子(Zn4,Zn6)分别被两个N2O2空腔包围,均是五配位的三角双锥空间构型.其中,Zn4原子与配体提供的两个肟氮原子(N5,N6)、两个酚氧原子(O14,O17)以及双齿桥联醋酸根中的一个氧原子(O23)配位;Zn6原子坐落在另外一个空腔内(N7,N8,O18,O21),同时与双齿桥联醋酸根中的一个氧原子(O25)配位.而中心的Zn(II)原子(Zn5)是六配位的,与配体中两个完全去质子的酚氧原子(O17,O18)、两个水分子的氧原子(O26,O27)和双齿桥联醋酸根上的两个氧原子(O22,O24)配位,形成扭曲的八面体空间构型(见图3(b)).
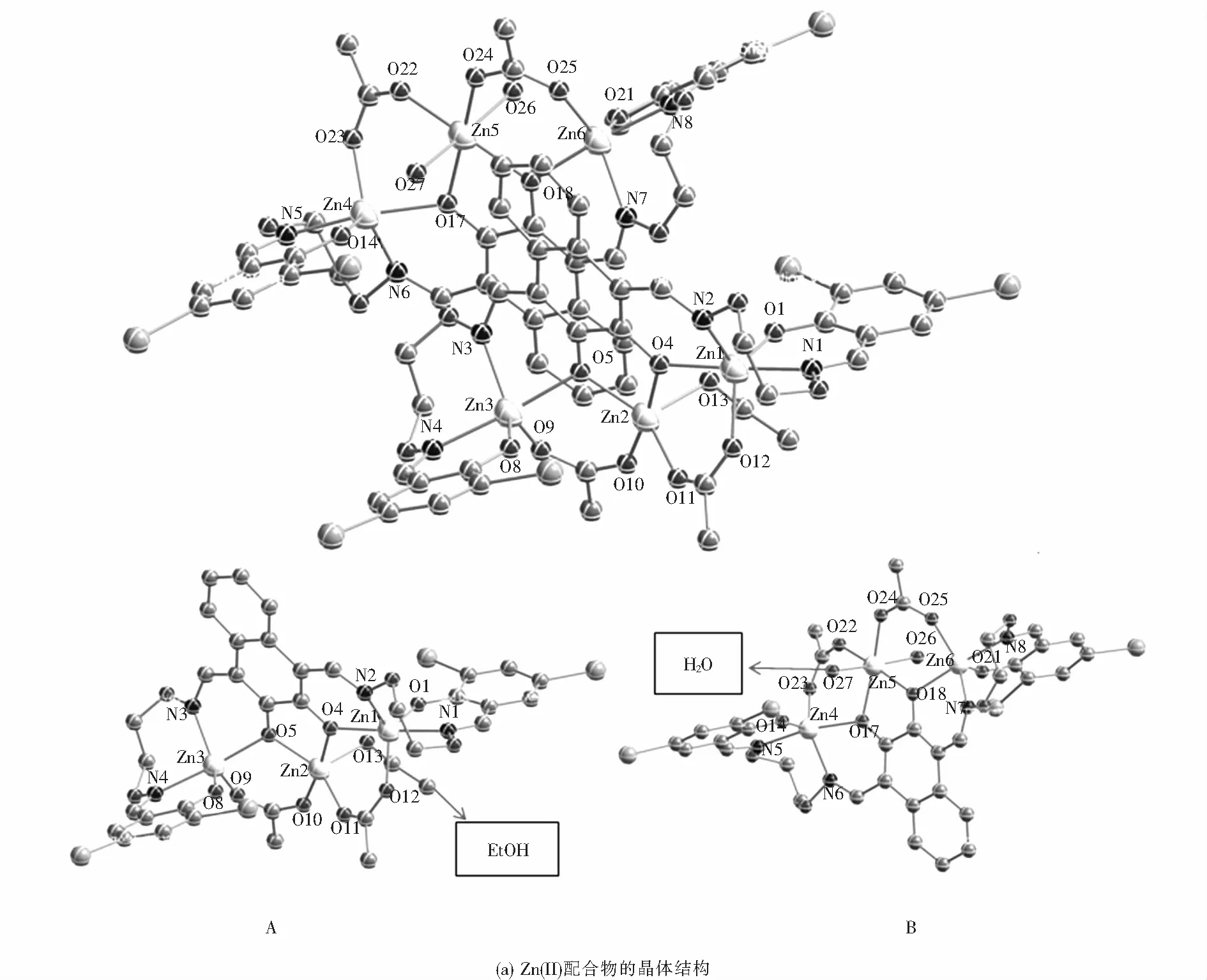

图3 Zn(II)配合物的晶体结构及Zn(II)原子的配位构型Fig.3 Crystal structure of Zn(II) complex and coordination configuration of Zn(II) atom
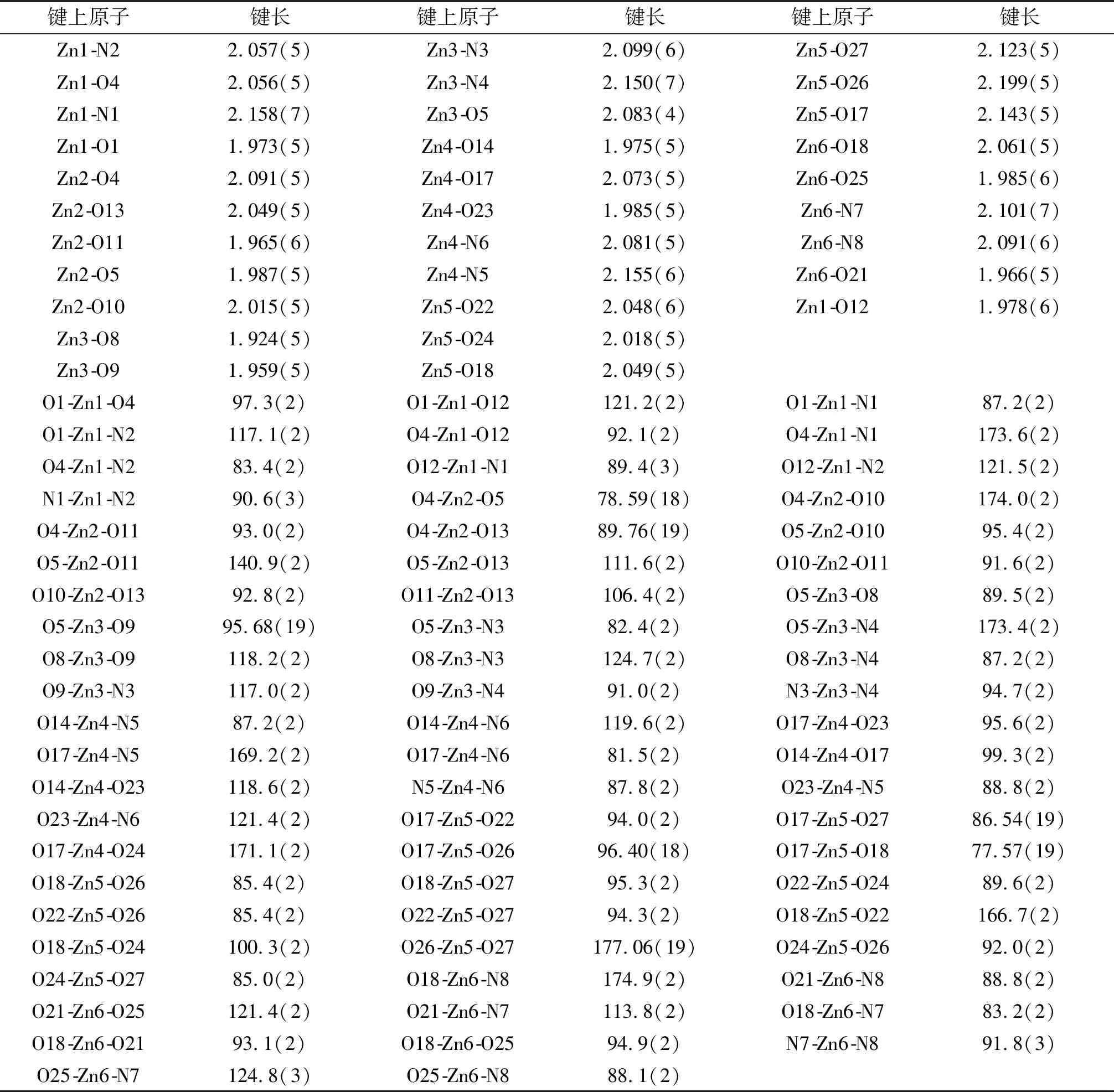
表6 锌(II)配合物的主要键长和键角
2.5 Zn(II)配合物的超分子作用分析
如图4(a),4(b)所示,配合物中有十一对分子内氢键相互作用(O13-H13…Br2,O13-H13…O1,O26-H26A…O21,O26-H26B…O22,O27-H27A…O24,O27-H27B…Br5,O27-H27B…O14,C8-H8B…O12,C23-H23B…O9,C44-H44A…O23和C59-H59A…O25),另外配合物还有四对分子间氢键相互作用(C9-H9B…O15,C30-H30…O3,C60-H60…O10,C60-H60…O11).通过这些分子间氢键沿着确定(bc)方向的相互作用形成结构稳定的二维(2-D)超分子结构.除此之外,如图4(c)所示,Zn(II)配合物分子通过分子间氢键相互作用形成了无限三维(3-D)超分子结构.Zn(II)配合物的氢键相互作用列于表7中.
2.6 Zn(II)配合物的Hirshfeld表面分析
通过Crystal Explorer 程序对Zn(II)配合物进行Hirshfeld表面分析,结果如图5所示,显示了dnorm、de和di映射上电子云分布情况.图中箭头所示不同区域分别表示配合物中O…H相互作用和C…H/H…C,H…H等相互作用.
图6为Hirshfeld表面分析二维指纹图,完整指纹的轮廓显示为灰色,各分图中的黑色区域代表不同的短程相互作用.通过计算得出配合物中C…H/H…C、O…H/H…O、H…H/H…H和Br…H/H…Br相互作用的占比分别为17.7%、8.1%、38.6%和21.4%,正是由于这些短程相互作用使Zn(II)配合物稳定存在.
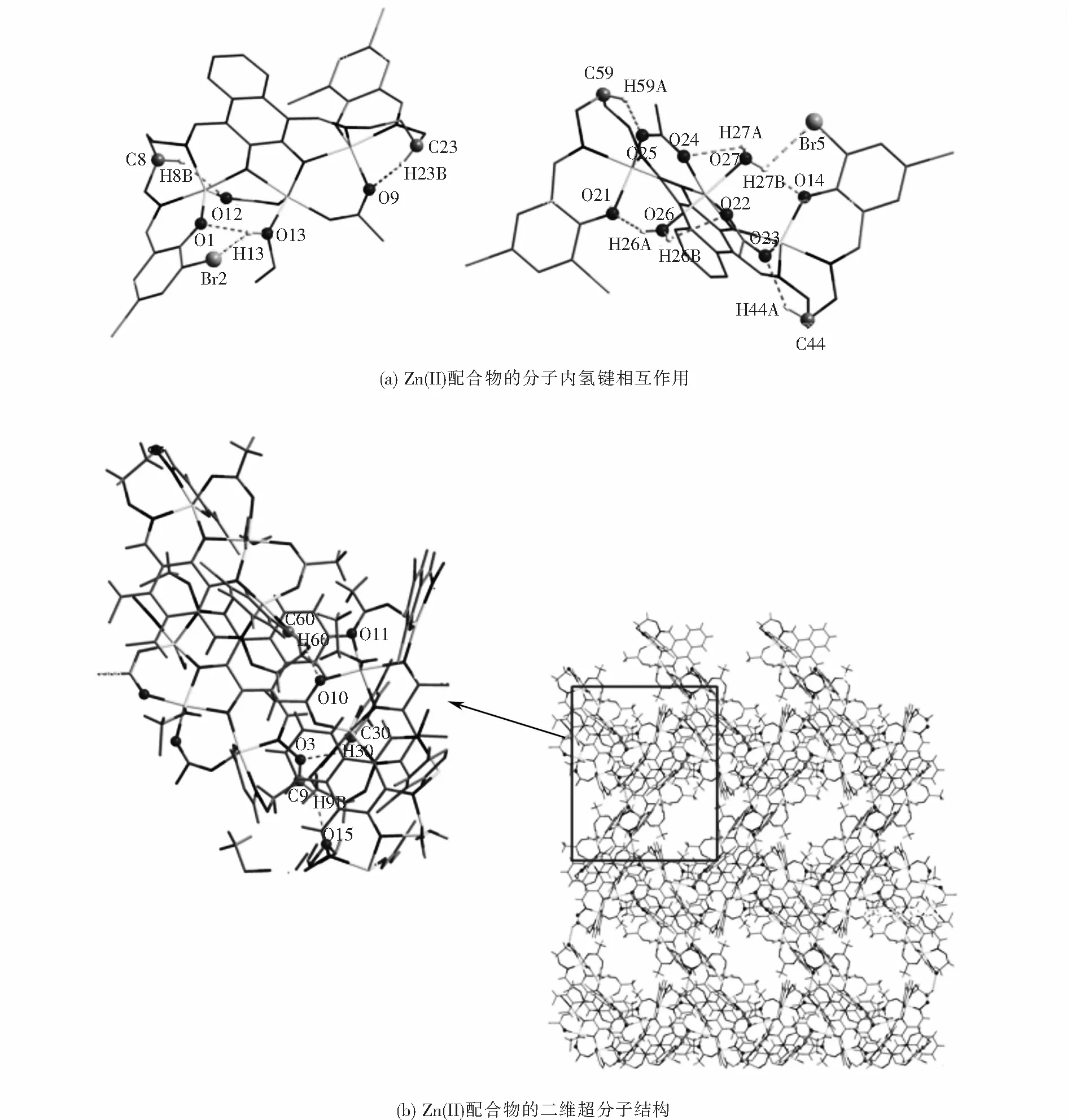
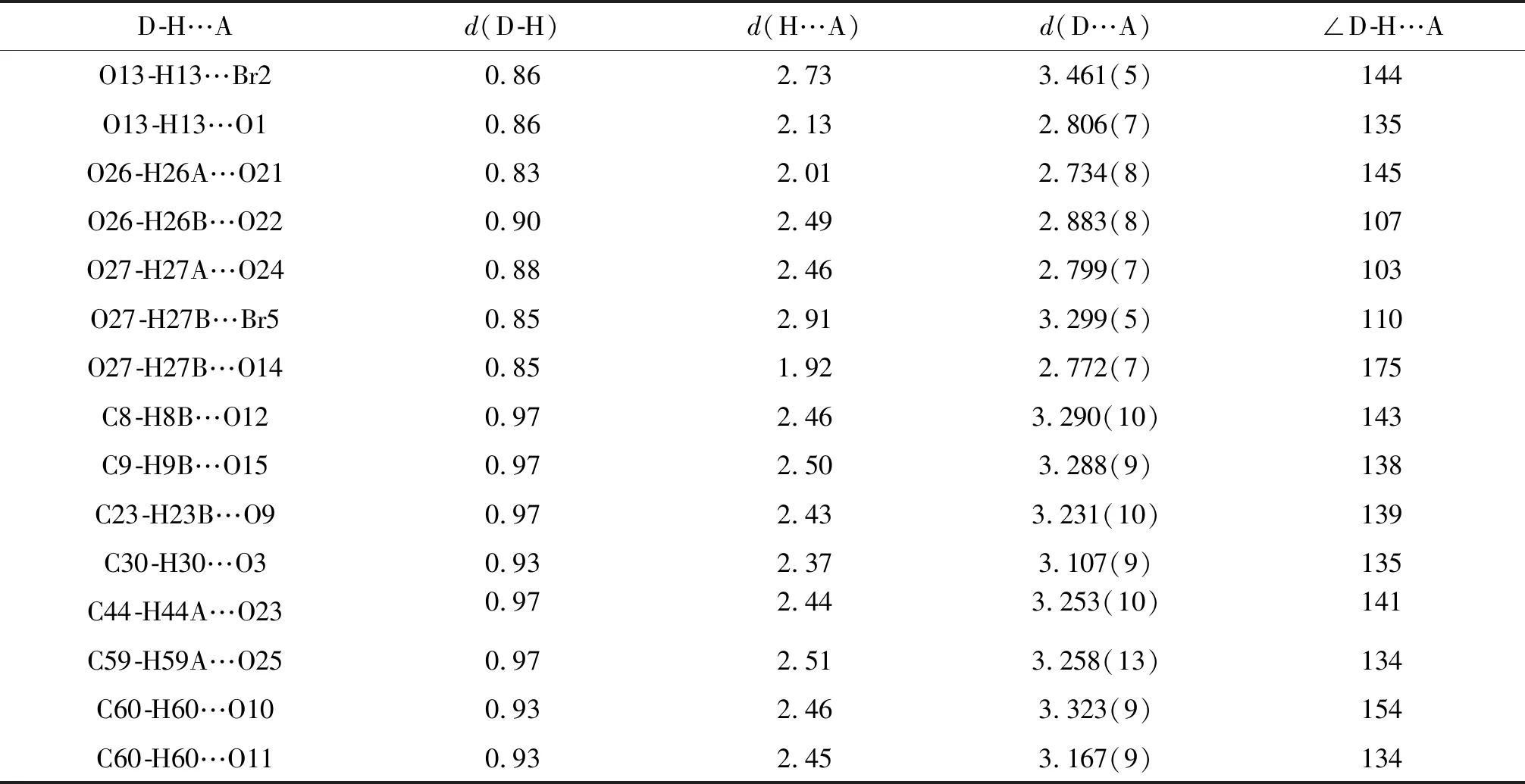
表7 Zn(II)配合物的分子内及分子间氢键的键长及键角
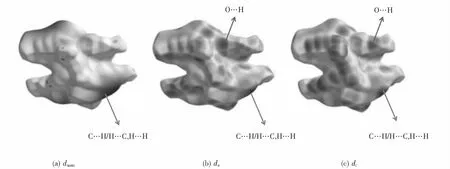
图5 Hirshfeld表面分析Fig.5 Hirshfeld surface analysis
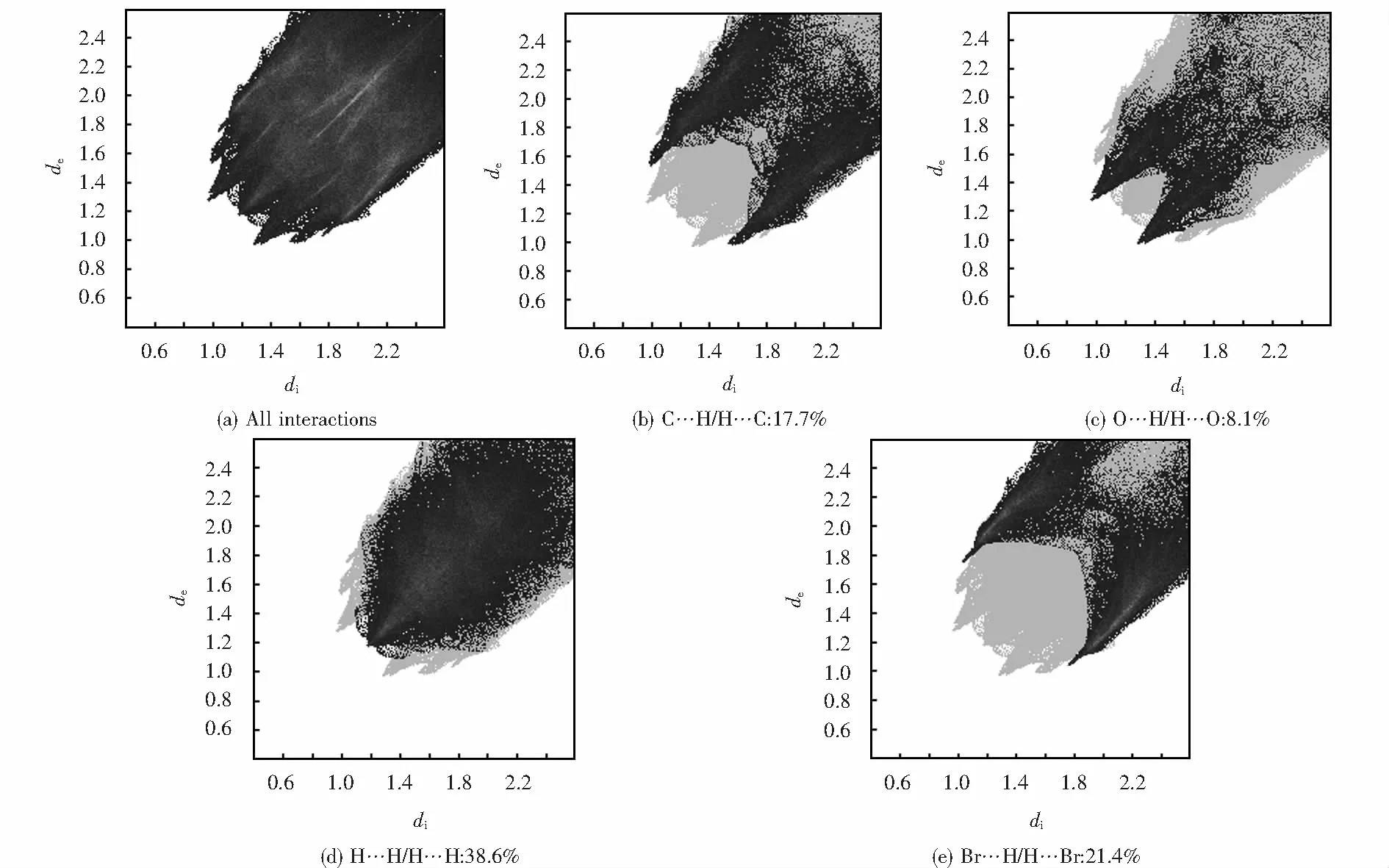
图6 Zn(II)配合物Hirshfeld表面接触百分比指纹图Fig.6 Fingerprint of Hirshfeld surface contact percentage of Zn(II) complex
2.7 Zn(II)配合物的DFT理论计算
为了深入研究Zn(II)配合物的电子结构,使用Gaussian-09软件对Zn(II)配合物进行DFT计算,通过密度泛函理论计算了分子轨道的能级差.Zn(II)配合物的分子轨道如图7所示,配合物的分子轨道能级可从A、B两部分分析,分别对应乙醇参与配位的独立单元和水分子参与配位的独立单元.乙醇参与配位的A独立单元中,HOMO分子轨道电子集中分布在3,5-二溴水杨醛上,LUMO分子轨道集中分布在Zn(II)原子和萘环上.分子中能量EHOMO=-5.655 2 eV,ELUMO=-2.530 1 eV,两个分子轨道的能极差(ΔE=ELUMO-EHOMO) 是3.125 1 eV.水分子参与配位的B独立单元中,存在α和β两个自旋分子轨道,α自旋轨道中EHOMO=-5.427 2 eV,ELUMO=-2.365 2 eV,能极差ΔE=3.062 eV;β自旋轨道中EHOMO=-5.732 5 eV,ELUMO=-2.588 9 eV,能极差ΔE=3.143 6 eV.ΔE值较小,表明配合物稳定性高.结果表明,在Zn(II)配合物的形成过程中,电子发生跃迁,形成了稳定性更高的配合物.
2.8 荧光光谱分析
图8显示了在1.0×10-3mol/L的DMF溶液中测定的配体H4L及其相应的Zn(II)配合物的荧光光谱.荧光测定参数值为激发波长412 nm,激发波长和发射波长的狭缝宽度均为10 nm,配体H4L在501 nm处出现一个较弱的发射峰,把它归属为苯环中的的π-π*跃迁.而Zn(II)配合物在此波长下激发时,490 nm处产生了一个强烈的发射峰,相对配体蓝移了11 nm,这可归因为配体到金属离子的电荷转移(LMCT)[23],说明Zn(II)离子与配体H4L配位形成了配合物.其荧光增强机理是由于具有d10电子构型的Zn(II)离子与配体H4L中的N2O2基团配位形成螯合环,增大了分子共轭程度,降低分子振动,从而导致配合物刚性增强,产生螯合荧光增强效应(CHEF)[18].
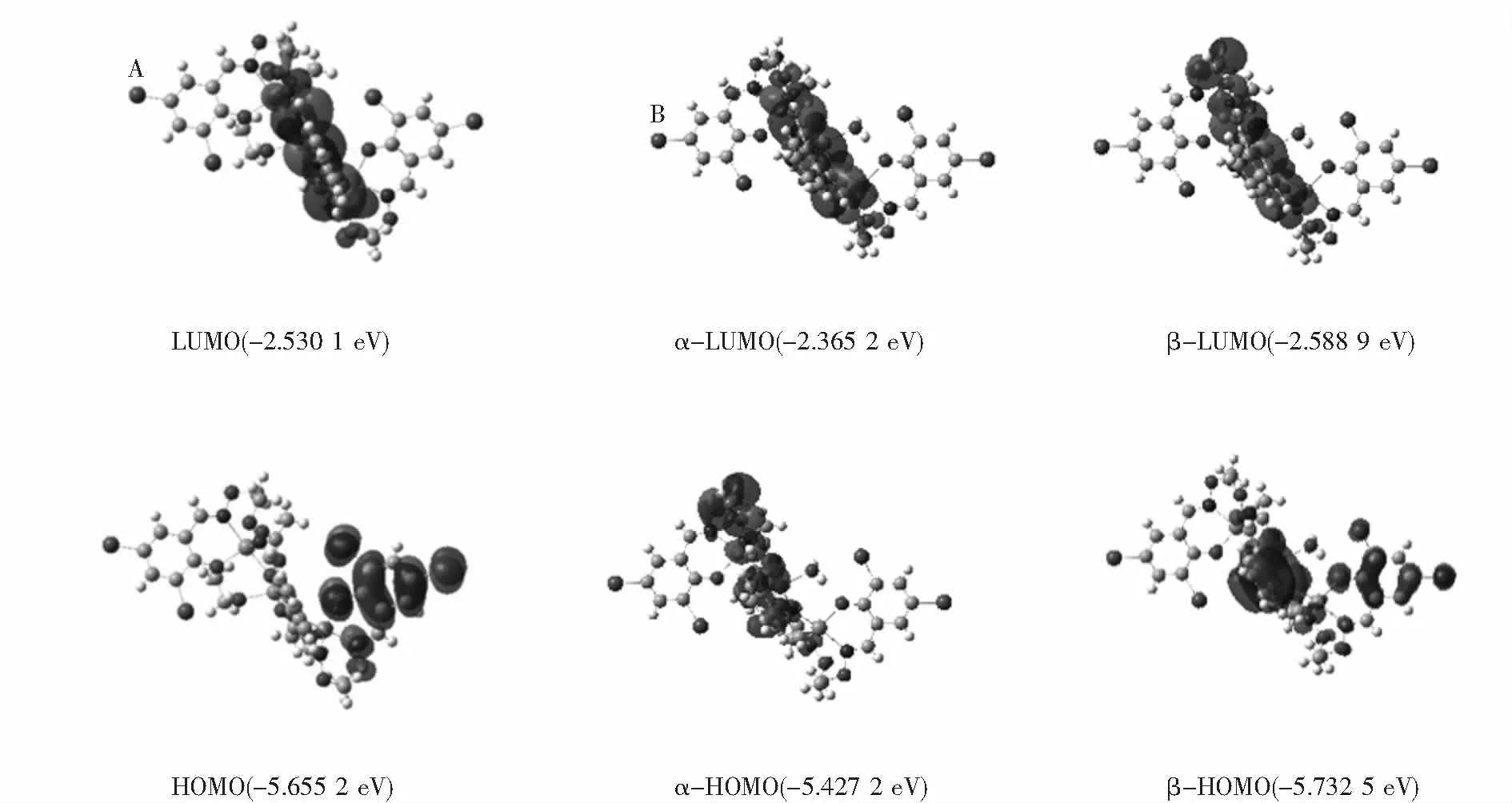
图7 Zn(II)配合物的分子轨道图Fig.7 Molecular orbital diagram of Zn(II) complex
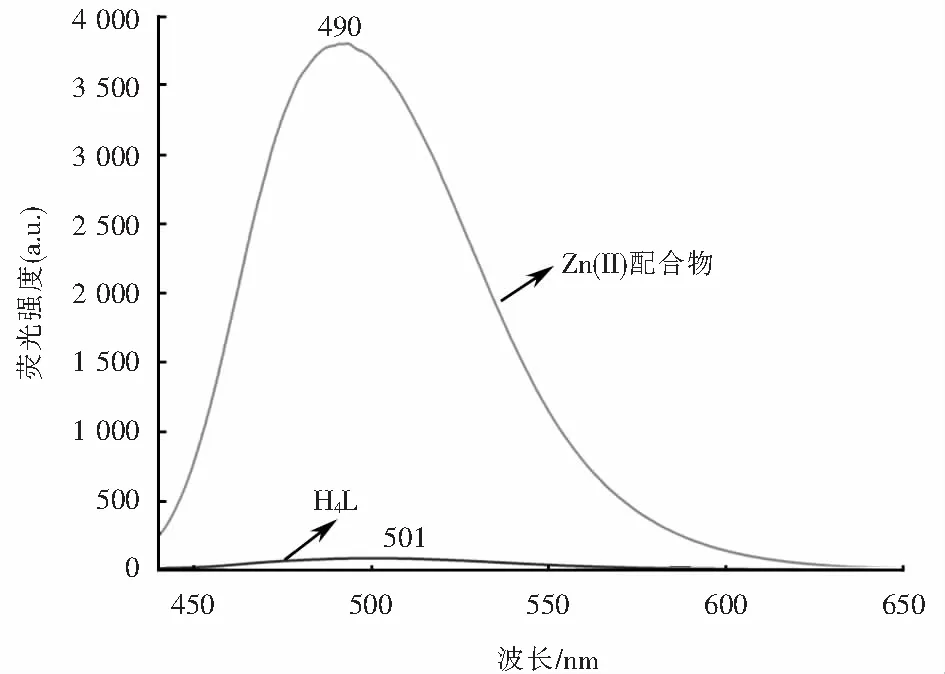
图8 配体H4L和Zn(II)配合物的荧光光谱Fig.8 Fluorescence spectra of ligand H4L and Zn(II) complex
3 结论
本文合成出了一种新的四溴代双Salamo型配体H4L,通过溶剂自然挥发法得到该配体与Zn(OAc)2·2H2O反应后的六核Zn(II)配合物单晶:[Zn3L(AcO)2(EtOH)][Zn3L(AcO)2(H2O)2],利用多种测试方法对配体H4L及其Zn(II)配合物进行了结构分析和性质表征.结果发现,该配合物在不同溶剂中溶解性差异大;红外光谱分析表明配体 H4L的N2O2配位环境有利于Zn(II)原子与之配位;晶体结构分析发现该配合物含有两个不同的配位单元,两个配位单元的相同之处是均含有三个Zn(II)原子、一个去质子化的(L)4-单元、两个桥联的醋酸根离子,不同之处在于中心Zn(II)原子配位的溶剂分子,一个是乙醇分子,另外一个是水分子参与配位;Hirshfeld表面分析表明配合物分子间存在氢键作用以及不同原子间的短程相互作用;通过DFT理论计算研究进一步证实了配合物的结构稳定性;荧光光谱结果表明,在同一激发波长下,当配体形成Zn(II)配合物后,荧光发射峰明显增强,其机理为螯合荧光增强效应(CHEF).以上工作都为环境中Zn(II)离子的识别与定量检测提供基础理论依据,对双Salamo型配合物的进一步研究具有指导意义.
