一例X-连锁无丙种球蛋白血症患者BTK基因新变异体的鉴定
2021-04-16王蓉蓉
张 晗,孙 阳,王蓉蓉,张 文,张 学*
(1.中国医学科学院基础医学研究所 北京协和医学院基础学院 麦库西克-张孝骞协和遗传医学中心医学分子生物学国家重点实验室,北京 100005;2.中国医学科学院 北京协和医学院 北京协和医院 风湿免疫科国家皮肤与免疫临床医学中心 卫生部重点实验室,北京 100730)
先天性无丙种球蛋白血症(congenital agammaglobulinemia)是一种罕见的原发性免疫缺陷病,其特征是血清免疫球蛋白水平低下和外周血B细胞缺乏。该病常呈X-连锁隐性遗传,但也有常染色体隐性遗传和常染色体显性遗传形式的报道。其中BTK变异导致的X-连锁无丙种球蛋白血症(X-linked agammaglobulinemia, XLA; OMIM 300755)占先天性无丙种球蛋白血症的85%。1952年Bruton最早报道XLA,故又被称为Bruton病[1-2]。美国XLA患者登记的结果显示,该病的最低发病率约为1/37.9万例活产(1/19万例男婴)[3]。中国目前尚无XLA患病率或发病率的准确估计。XLA是由于位于Xq22.1上的Tec家族成员Bruton酪氨酸激酶(Bruton tyrosine kinase, BTK)基因发生变异导致的一种原发性体液免疫缺陷病,是典型的原发性B细胞缺陷病[4-6]。BTK基因变异会使得B细胞系发育障碍,血清免疫球蛋白合成不足,最终导致患者感染易感性增加[3, 7-8]。
本研究结合临床表型、全外显子组测序技术(whole exome sequencing, WES)、Sanger测序和实时定量PCR,通过相关基因诊断技术和遗传学分析,对一例XLA患者进行研究,为该病的基因诊断、遗传咨询以及治疗提供依据。
1 材料与方法
1.1 材料
1.1.1 研究对象:男性,31岁,由于3个月内反复发热到北京协和医院风湿免疫科就诊。在获得患者及其家庭成员的知情同意后,收集患者及其家系成员的临床资料、家族史并对患者进行临床诊断及家系分析(图1)。该研究遵循赫尔辛基宣言(Helsinki Declaration),并获得中国医学科学院基础医学研究所伦理委员会的审查批准(审批文号:015-2015)。

The square represents male, the circle represents female, the black symbol represents illness, the proband is indicated by an arrow图1 X-连锁无丙种球蛋白血症家系图谱Fig 1 Pedigree diagram of the family with X-linked agammaglobulinemia
1.1.2 试剂:DNA提取试剂盒(QIAGEN公司);引物合成(北京天一辉远生物科技有限公司)Trizol试剂盒(Invitrogen公司);反转录试剂盒(TaKaRa公司)。
1.2 研究方法
1.2.1 样本的采集与DNA的提取:留取患者及父母的外周血样本并用全血细胞DNA提取试剂盒提取样本基因组DNA。
1.2.2 WES测序筛选变异:取患者外周血基因组DNA 1 μg送至北京诺禾致源科技股份有限公司进行WES检测和数据分析,使用HiSeq 2500 PE125测序系统(美国Illumina公司)进行人类全外显子组测序。并按照如下策略进行分析和筛选:1)按照遗传方式关注呈X-连锁遗传和常染色体隐性遗传的变异,不排除新生变异;2)选择最小等位基因频率≤0.001的变异;3)关注位于基因编码区且改变所编码蛋白质的变异及可能改变mRNA剪接的变异,如非同义变异、移码变异和剪接位点变异;4)针对候选基因已报道的基因功能及软件对变异的有害性进行预测,得到候选基因变异体;5)参考2015年美国医学遗传学和基因组学学会(ACMG) 针对序列变异解读的标准和指南,对所得到变异的有害性进行分类,关注分类为致病、可疑致病和意义不明确的变异。
1.2.3 Sanger测序验证:针对WES筛选出来的变异位点设计引物,从UCSC基因数据库(http://genome.ucsc.edu/)获取基因序列信息,利用Primer3 Input(version 4.1.0)(http://primer3.ut.ee/)进行在线设计引物,由北京天一辉远生物科技有限公司合成。PCR扩增患者及父母DNA样本后,扩增产物送北京诺赛基因公司进行测序。
1.2.4 RNA的提取及cDNA测序:用Trizol试剂盒提取患者及其父亲外周血中总RNA,使用反转录试剂盒反转录2 μg RNA获得cDNA。PCR扩增患者及其父亲cDNA样本后,扩增产物送北京诺赛基因公司进行测序。
1.2.5 实时定量PCR检测mRNA:以β-Actin为内参,使用荧光定量PCR仪(QuantStudio®3 Real-Time PCR Instrument)检测BTKmRNA的相对表达量。数据采用2-△△Ct法分析,具体方法为:△Ct=目标基因Ct均值-内参基因Ct均值,△△Ct=患者△Ct-健康对照△Ct。实时定量PCR实验重复3次。
2 结果
2.1 临床信息
患者(图1)男性,31岁,因反复发热、咳嗽就诊。自诉自幼经常发热,并伴有反复的上呼吸道感染及肺部感染,咳嗽、咳痰,否认痰中带血。既往患有慢性支气管炎。患者因无明显诱因下经常出现腹痛、水样便腹泻被诊断为“慢性结肠炎”,否认便血。偶发双膝关节疼痛。患者生长曲线正常。患者父母否认近亲婚配史,患者否认其男性亲属有类似疾病。母亲妊娠期间无异常。
2.2 实验室检查
患者血清免疫球蛋白IgA、IgM、IgG显著降低,IgE大致正常。患者外周血淋巴细胞亚群分析显示,外周血B淋巴细胞(CD19+)百分比为0,显著降低。自然杀伤细胞(CD16+CD56+)百分比轻微降低,外周血T细胞(CD3+、CD4+、 CD8+、CD4+/ CD8+)正常。
2.3 WES测序结果分析和Sanger测序验证
通过WES测序,总共得到10 G的数据量,平均深度>100*。通过上述分析流程,鉴定出患者在BTK(NM_000061)的3号内含子c.240+3A>C处有一变异,Sanger测序发现该变异来源于其母亲,符合家系基因型-表型共分离(图2)。该变异在dbSNP153数据库、ExAC数据库、gnomAD数据库以及人类基因突变数据库 (Human Gene Mutation Database, HGMD)中均未见报道。
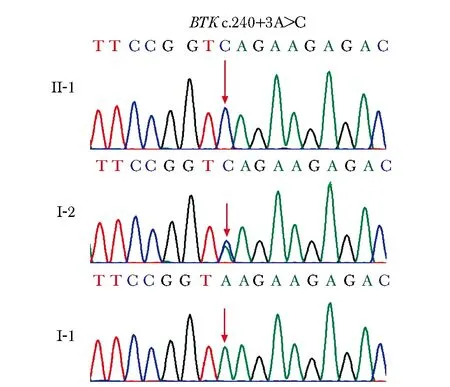
图2 患者及其父母BTK变异位点测序图Fig 2 Sanger sequence chromatograms of the BTK variant in the patient and his parents
2.4 cDNA测序和mRNA定量检测
cDNA测序显示c.240+3A>C变异BTK3号内含子5′端106 bp碱基插入到BTK转录本第3号和第4号外显子之间,引起阅读框改变,终止密码子提前出现(图3)。实时定量PCR显示,与无血缘关系正常男性(control)及其父亲(Ⅰ-1)相比,患者(Ⅱ-1)BTK的mRNA表达水平明显降低(图4)。
3 讨论
先天性无丙种球蛋白血症是一种由于B细胞早期发育障碍所致的原发性免疫缺陷病,包括X-连锁隐性遗传、常染色体隐性遗传和常染色体显性遗传。其中,XLA是一种由于B细胞早期发育障碍所致的原发性免疫缺陷病,呈X-连锁隐性遗传。患者多于6到12个月龄时起病,其最突出的临床特征为早发、 反复的严重细菌感染[9]。XLA感染部位主要包括呼吸系统和消化系统,另外,还容易出现中耳炎、慢性鼻窦炎、皮肌炎等并发症[6]。因该患者有反复的感染史,实验室检查结果显示血清免疫球蛋白减少,外周血成熟B淋巴细胞缺如,怀疑其患有先天性无丙种球蛋白血症,因此需对患者进行基因检测辅助临床诊断。

A.sanger sequence chromatograms of the BTK cDNA; B.schernatyc presentation dragram of BTK cDNA
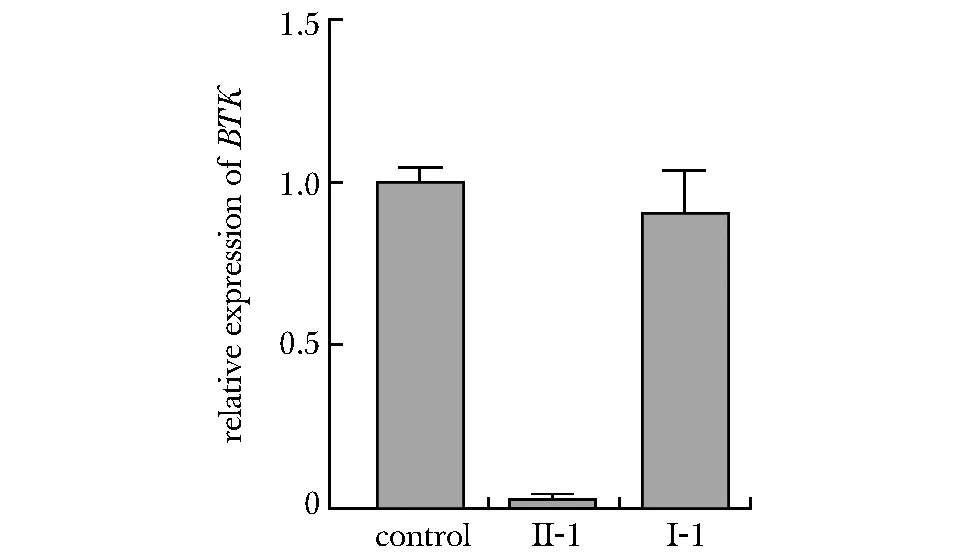
The values presented are means of triplicate determinations,the control was set to 1.0,the Y-axis represented the relative expression of BTK, the results of the experiment were repeated three times图4 患者及患者父亲BTK相对表达量Fig 4 Relative mRNA expression of the BTK in the patient and his
BTK变异分析是XLA的诊断依据。BTK基因位于Xq22.1,长度为37 kb,包含有19个外显子。该基因编码的细胞质酪氨酸激酶蛋白(BTK)含有5个功能结构域,分别为PH、TH、SH2、SH3和TK[10-11],已报道的变异主要包括错义变异、无义变异、插入或缺失变异和剪接变异等,暂未发现突变热点区域。其分子缺陷会导致B细胞系列发育障碍和功能障碍,从而导致免疫球蛋白缺乏和特异性抗体产生障碍[11-12]。本研究中患者携带c.240+3A>C变异,HGMD中未见报道,该突变导致来自3号内含子5′端的106 bp碱基插入到BTK转录本3号外显子和4号外显子之间,提前出现终止密码子,mRNA发生降解,最终导致患者XLA疾病的发生。
BTK变异体检测有助于XLA 早期诊断。全外显子组测序技术的快速发展和广泛应用,有助于BTK变异的鉴定。XLA如果不正规治疗将会导致该病发病率和病死率激增[3]。早期诊断并给予有效治疗有助于减少患者反复的细菌感染,避免严重并发症的发生,改善患者的生存质量。自1952年Bruton成功地使用替代免疫球蛋白疗法治疗了第一例患者以来,免疫球蛋白替代疗法一直是XLA的主要治疗方法,目前有静脉注射免疫球蛋白(intravenous immunoglobulin, IVIG)途径和皮下注射免疫球蛋白(subcutaneous immunoglobulin, SCIg)途径两种[1, 13-14]。
综上所述,本研究结合患者临床表型、实验室检查结果以及患者遗传学检测结果,鉴定了一个XLA的致病基因变异,为该患者的基因诊断、遗传咨询以及治疗提供了依据;发现了BTKc.240+3A>C这一新的变异位点,丰富了BTK基因变异数据库。
