MgO/Mg(OH)2热化学储热反应的第一性原理研究
2021-04-09王琴徐会金韩兴超赵长颖
王琴,徐会金,韩兴超,赵长颖
(上海交通大学中英国际低碳学院,上海200240)
引 言
据BP《世界能源统计年鉴2019》公布的2018年世界能源消费统计,全球一次能源消费近十年增长率均值为1.5%,在2018 年几乎达到了两倍(2.9%),全球能源消费量达到138.65 亿吨油当量。其中,煤炭消费增长1.62 亿吨油当量,增长率1.4%,石油消费量增长140 万桶/日,增长率1.5%,天然气消费增长1950 亿立方米,增长率5.3%[1-2]。如今,即使在大力发展可再生能源,化石燃料依然是主要的能量来源,可再生能源的占比仍相对较低。由于经济社会的高速发展,能源需求加大,化石燃料大量燃烧,给生态环境带来了巨大的压力,因此,加速推动能源系统低碳转型仍然十分必要。在能源结构的持续改进过程中,储能也扮演着越来越重要的角色,它可以解决由于时间、空间或强度上的热能供给与需求间不匹配所带来的问题。对比储热技术中的显热储热、潜热储热,热化学储热储能密度最大[3-4],且其具有储能密度高、储热周期长、常温下可存储储能物质的分解物并实现其远距离传输等优点[5-6]。
在热化学储热反应中,热量是通过反应焓来存储的,脱水反应为蓄热过程,水合为放热过程。MgO/Mg(OH)2体系是一种很有前途的热化学蓄热方法,其反应方程[7]为:
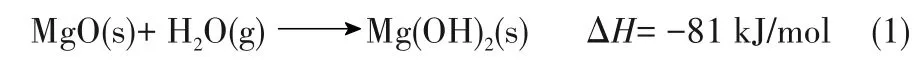
在热化学储热材料中,氧化镁是最便宜的一种,且安全无毒,其储热能力也相当大(约3.3 GJ/m3)。此 外,MgO/Mg(OH)2系 统 反 应 温 度 在350~400℃之间,条件相对较低,在处理工业余热方面有很大潜力[8-12]。
目前,国内外对MgO/Mg(OH)2热化学储热体系的应用研究主要集中在反应物体系热力性质的优化、循环性能测试、储热系统的设计及测试、反应器强化传热传质等方面[13]。Nahdi 等[14]利用控制速率热分析技术测试了MgO/Mg(OH)2水合/分解特性,并进行了一系列典型的材料表征。Kato等[15]将Mg(OH)2和Ni(OH)2混合制成MgxNi1-x(OH)2(x为混合氢氧化物中镁离子占总金属离子的摩尔分数)并进行了一些列性能测试,发现混合Ni(OH)2可将反应温度降低至200~300℃,且可以通过改变材料中混合金属氧化物的组成来提高操作蓄热温度。Mastronardo 等[7]提出了一种新型氢氧化镁和碳纳米管复合材料作为MgO/H2O/Mg(OH)2化学热泵的蓄热介质,并采用热重分析法对合成材料的性能进行了评估,结果表明碳质材料的存在对反应性能有积极影响,使杂化材料比纯Mg(OH)2具有更好的蓄热/产热能力和更快的产热速率。Haruki 等[16]制备氢氧化镁/氧化镁/膨胀石墨复合材料,测试了不同配比下其导热性和反应性。目前研究主要采用的是实验研究和特性表征,对储热系统的微观机理及微观因素如何影响到宏观储热的过程方面的深入研究还比较缺乏。虽然在分子动力学有部分的研究,但是针对氧化镁的储热机理、掺杂盐中具体哪种原子对储热系统有何种程度的影响的研究仍较少,所以基于第一性原理对这方面的研究是很有必要的。在其他研究领域,已有一些基于第一性原理对某些微观机理的研究。Ding等[17]对第一性原理在催化机理研究中和吸附过程中的应用进行了归纳总结,发现第一性原理在理论分析中有很大的优势,在原位模型催化剂表面模拟计算中效率突出。然而现有研究仍较少涉及储热系统的微观机理。Carrasco 等[18]用第一性原理和ab initio 分子动力学模拟的方法研究了碱土氧化物(如CaO、MgO、BaO)对低覆盖水的吸附,结果表明,MgO(001)表面质子-羟基结构在单步形成和分离过程中存在较大的障碍,而CaO 和BaO(001)表面羟基可以围绕固定的质子自由旋转,使其具有更好的水解离特性。Wang 等[19]利用第一性原理方法研究了MgH2中与氢有关的固有缺陷和与F 或Cl 有关的杂质的结构特性和形成能,结果表明,在带电荷状态下,氢空位和间隙是有利的,而F和Cl杂质倾向于以中性状态取代氢原子;F 和Cl 对中性和带负电荷的H 原子的去除能量均无影响,但对带正电荷的H 原子去除能量均有显著影响。刘亮等[20]采用第一性原理计算研究了碱金属和碱土金属对CaO(100)理想表面和促进表面的吸附,结果表明,CO2倾向于吸附在O-top位点上,形成最稳定的构型,Na、K、Rb和Cs因其较高的电正性而被认为是九种金属中合适的促进剂。申洁等[21]采用基于密度泛函理论的第一性原理赝势平面波方法计算Eu 原子掺杂MgO 晶体的几何结构、能带结构、态密度和光学性质,计算结果表明掺杂稀土元素对体系的静态介电常数、晶体反射率、光吸收系数、折射率和能量损失函数有重要调制作用。可发现,目前研究主要集中于反应物晶体缺陷、晶体构型等方面,对氧化镁的热力学性质和最稳定微观吸附构型研究较少。
针对MgO/Mg(OH)2储热系统,本文根据已有的类似微观研究,基于密度泛函理论的广义梯度近似,对H2O 在MgO 表面的吸附进行了模拟计算,以此来探究氧化镁的储热机理。此外,也就掺杂盐对材料改性进行了深入的探索,探究掺杂盐中的金属原子在提升储热性能方面的影响。通过计算找到最符合实际的微观吸附构型,在原子分子水平上深入认识了H2O 与MgO 的反应过程。在掺杂盐性能影响研究中,本文针对最稳定构型进行Li、Na、K 三种金属原子掺杂,研究了其活化能和热力学性质,并进行了对比分析,从微观层面探索金属原子对储热的影响效果,这对后续储热系统研究及其开发应用有较好的参考价值。
1 计算方法和模型
1.1 计算方法
本文吸附和吸附构型计算均采用基于密度泛函理论的CASTEP(Cambridge Serial Total Energy Package)程序包进行第一性原理研究[22],掺杂计算采用Dmol3程序包,吸附计算中泛函和极化双数值轨道基组采用广义梯度近似(general gradient approximate,GGA)中的PBE 处理交换关联能。平面波截断能设置为489.8 eV,布里渊区K 点取样为6×6×6。自洽场(SCF)收敛于2.0×10-5eV/atom,最大迭代次数500,多极展开到6 极,Density Mixing 方法中charge 和spin 分别为0.2 和0.5,DIIS 外推大小为6。热力学性能计算中泛函和极化双数值轨道基组采用局部密度近似(local density approximation,LDA)中的CA-PZE 处理交换关联能。平面波截断能设置为990.0 eV,K 点取样为6×6×6,Pseudo-potentials 设置为Norm-conserving。
在模拟计算之前,需要对分子/晶体结构进行优化,使结构与实际分子/晶体结构相吻合,再进行模拟计算,这样模拟的结果更符合实际,更具有指导性意义。首先采用这些计算参数对H2O及晶体MgO的结构参数进行优化测试计算,以确定平衡态原子的精确位置: H2O 的O—H 键长是0.976 nm,角度为104.727°,优化计算结果如图1 所示。与文献[23]中H2O 的O—H 键长0.971 nm、角度为104.5°实验值相比,键长偏差为0.51%,角度偏差为0.22%;而MgO的晶格常数计算值为aMgO=4.21120 Å(1 Å=0.1 nm),与文献[24-27]具有很好的一致性,图2展示了本研究建立的氧化镁模型的微观计算参数与四篇文献中的实验数据对比结果,模型与实验数据偏差较小,有很好的一致性。
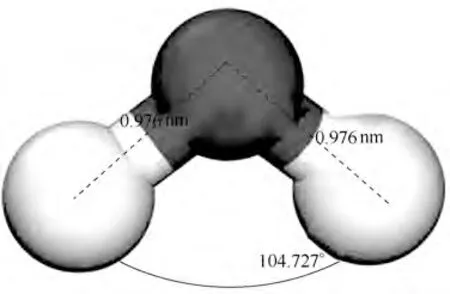
图1 优化后的水分子结构(白色为H,黑色为O)Fig.1 The optimized water substructure(white is H and black is O)
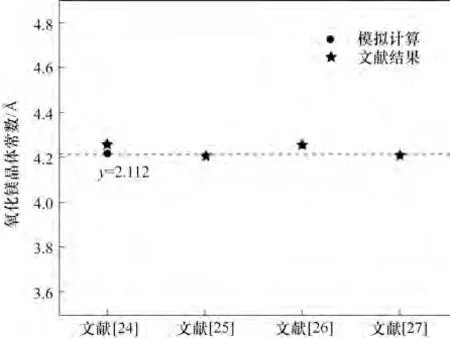
图2 氧化镁微观参数计算模型的验证Fig.2 Verification of the microscopic parameters of MgO model
1.2 计算模型
1.2.1 氧化镁结构模型 氧化镁是典型的立方晶系结构——NaCl 构型,空间点群为Fm-3m(No.225),一个氧化镁单胞中包含4 个O 原子和4 个Mg 原子,分别占据Wyckoff 坐标(表示晶胞中等价原子的对称性,包括多重度、位置的对称性以及该位置的分数坐标)的4a 和4b 位置。图3(a)为传统的MgO 晶胞,显示了晶格的立方对称性。利用晶格的对称性,使用只包含2 个原子的原胞[图3(b)]来进行热力学性质计算(而晶胞包含了8 个原子),得到的电荷密度、键长等与晶体直接计算结果一样,但是计算量大大减小,计算时间被缩短。
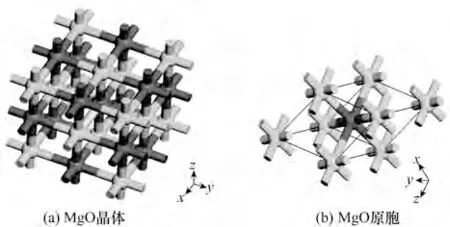
图3 MgO晶体结构(深色为O,浅色为Mg)Fig.3 Crystal structure of magnesium oxide(dark is O and light is Mg)
1.2.2 吸附模型 理想的氧化镁表面没有任何杂质和缺陷,但实际上很少见。实际中考虑最多的表面畸变是表面弛豫和表面重构。为了研究孤立水分子在氧化镁表面的性质,本文采用表面层相对于理想表面原子晶格运动但表面原胞对称性不改变的表面弛豫进行研究。首先,建立氧化镁超晶胞结构:对建立的MgO 晶体结构进行几何优化,基于Carrasco 等[18]的研究,整个计算过程主要在MgO 的(001)面进行,所以对(001)表面进行切割,如图4 所示。所有表面的原子层数均为3 层,为保证原子层间不发生相互作用,z 方向周期性排列的slab 之间有1.5 nm 的真空区域,因为超晶胞的对称性,本文以侧视图和俯视图展示,相关结构如图5 所示。随后对采用周期性的三层结构(Mg12O12)的超晶胞模型进行全弛豫优化,以确定平衡态的原子精确位置。
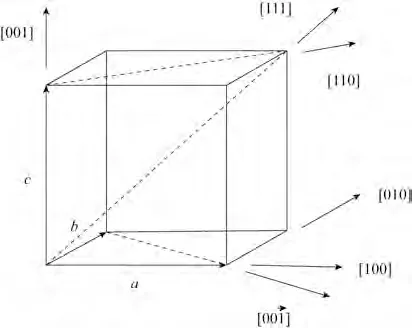
图4 氧化镁立方晶体晶列指数[28]Fig.4 Index of cubic crystal of magnesium oxide[28]
活性组分在MgO 表面的吸附能Eads定义为吸附前后总能量的变化:
Eads= EM/slab-(EM+ Eslab) (2)
式中,EM/slab为活性组分在MgO 表面吸附后的能量;EM和Eslab分别为活性组分自由原子与表面的能量。
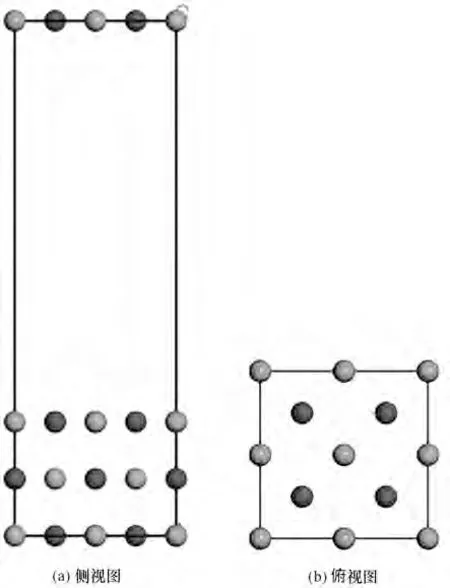
图5 P(2×2)超晶胞3层平板模型(深色和浅色分别为镁原子、氧原子)Fig.5 3-Layer supercrystal cell plate model(small dark spheres stand for Mg atoms,while light for O atoms)
根据该定义,吸附能为负值时表明活性组分的负载是放热过程,吸附能越小,活性组分在氧化镁表面的吸附作用越强烈,且吸附后体系更加稳定。Eads<0.62 eV(1 eV=96.4853 kJ/mol)时为化学吸附,反之为物理吸附[29]。
2 结果分析
2.1 分子与底物距离对吸附的影响
本研究采用P(2×2)超晶胞3 层平板模型,首先研究不同吸附距离对吸附反应的影响,图6 为H2O在MgO(001)表面吸附示意图,d 为分子距离底物(MgO)的距离,其取值分别为1.5、2.0、2.5、3.0 Å。
从表1可以看出,分子距离底物距离不同,吸附能也有所不同。其中距离为1.5 Å 时吸附能偏差最小,其次是距离为2.0 Å情况下,距离为3.0 Å的情况为吸附能偏差最大的距离。这是因为物理吸附和化学吸附共同作用。物理吸附是比较弱的吸附,吸附物和表面之间没有形成真正的化学键,没有电子共用,电子结构没有发生大的变化,通常物理吸附分为距离较近的强排斥力和距离稍远的相互吸引力;化学吸附是强吸附,吸附物和表面之间形成化学键有共用电子,它们的电子结构都有大的改变,涉及吸附分子和固体间的电子重排、化学键的断裂或形成。H2O 在MgO(001)表面的吸附能大于0.62 eV,属于化学吸附。
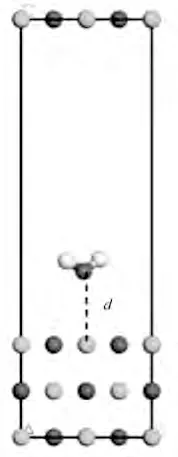
图6 H2O在MgO(001)表面吸附(白色为氢原子)Fig.6 H2O adsorbed on the surface of MgO(001)(small white spheres stand for H atoms)

表1 不同吸附距离吸附能对比Table 1 Comparison of adsorption energy for different adsorption distances
费米能级两侧分别有两个尖峰,尖峰之间的距离为赝能隙(pseudogap)[30],局部某个键处的赝能隙的宽度变化反映了此处键能的变化,赝能隙宽度增加意味着更强的化学键,反之,赝能隙变窄代表此处化学键更易断裂。O 的电子结构是2s22p4[31],Mg的电子结构是2s22p63s2,从四种不同距离的吸附表面态密度图(图7)可知,在一定距离内,态密度的轨道峰位置和赝能隙宽度基本没有变化,但当距离超过这个范围,轨道峰开始向左移动,赝能隙变小,轨道峰的面积呈减小趋势。其中,分子距离底物在0.3 nm 以内时,轨道峰几乎没有移动,-25~-5 eV 区间的轨道峰面积基本不变,而-5~15 eV 轨道峰面积略有所不同,但差别较小。距离底物大于0.3 nm时,轨道峰整体往左移动,-5~0 eV 区间和0~10 eV区间的轨道峰变高变窄,10~15 eV 对应的s 轨道峰几乎消失不见。反应态密度的积分为电子数,从图中的面积对比可以看出,位于最外层的p 轨道靠近费米能级的峰因为吸附距离变大面积变小,s 轨道也与此类似,说明距离拉远到一定时,电子转移受到距离限制而减少,即反应更难进行。
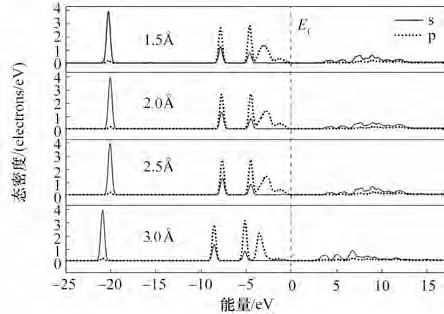
图7 不同距离吸附的表面吸附态密度分布Fig.7 Density of state at different distances
2.2 不同吸附构型对吸附的影响
对H2O 在MgO(001)表面存在的4 种空间吸附构型(镁顶位、氧顶位、二重桥位及四重洞位,如图8所示)进行比较。根据前文计算的结果,H2O 与底物的初始距离均设为最理想的吸附距离,分别对4 种初始吸附构型进行优化,结果如表2所示。
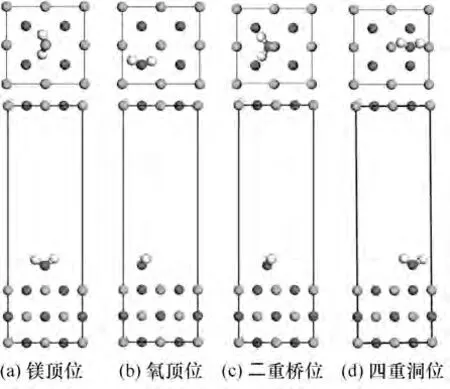
图8 H2O在MgO(001)表面4种吸附位点Fig.8 Four adsorption configurations of H2O on MgO(001)surface
表2 给出了4 种吸附位点下吸附能(Eads),结果表明,四重洞位的吸附能最低,说明H2O在这个位点的吸附不够稳定;而二重桥位和镁顶位的吸附能较大,分别达到了-0.8265 eV 和-0.8341 eV,为化学吸附,而氧顶位和四重洞位的吸附能均小于0.62 eV,为物理吸附。四种吸附构型中,镁顶位的吸附能最接近实验值(-0.8395 eV)。从距离上来看,O-Mg 距离处于Ha-Mg和Hb-Mg距离之间,说明H2O 中的氢氧根倾向于向Mg 原子方向移动,与表面的Mg 原子相连接,而另一个氢原子与表面的氧原子结合形成氢氧键。Ha-O 和Hb-O 距离差别很小,说明水分子仍然处于比较对称状态。其中,对于O-Mg 距离最远的为氧顶位,对于其他原子距离,也是氧顶位吸附位距离最远,说明氧顶位更不利于原子结合,即不易于吸附;∠Ha-O-Hb 角度氧顶位最大,四重洞位角度最小,说明水分子角度发生扭转,尽可能使氢原子贴近镁原子,更有利于吸附,氧顶位扭转最大,更贴近镁原子,更易于吸附。
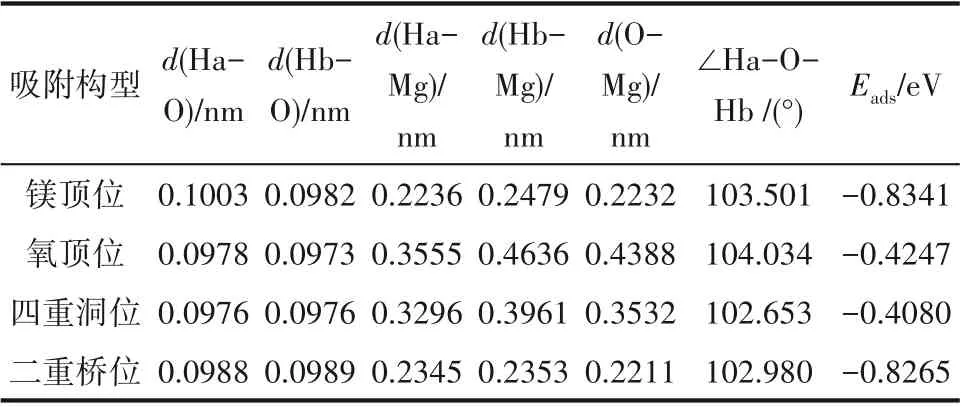
表2 H2O吸附于MgO(001)表面不同位点的稳定性比较Table 2 Comparison of the stability of the four adsorption configurations
图9 为H2O 在MgO(001)表面在4 种构型下吸附后的结构图,由图可以发现,二重桥位和镁顶位吸附前后与镁原子的距离变化较小,而氧顶位和四重洞位的距离变化较大。原因是初始位置时,MgO 中的氧原子与H2O 中的氧原子之间发生互斥,H2O 与氧化镁发生化学反应。吸附后稳定构型中的水分子都发生了扭转,扭转后水分子中的H 原子都转向了晶体表面,因为在形成化学键过程中,水分子中的H原子会发生断键,与晶体中的O原子结合,形成氢氧键。
图10 是H2O 在Mg 顶位吸附在MgO(001)表面的稳定构型,达到平衡时,H2O 中的氧原子和氧化镁中的镁原子距离为0.2232 nm,达到平衡后H2O 中的∠Ha-O-Hb 角度发生扭转,相比于初始构型,达到平衡后的∠Ha-O-Hb 转变为103.501°,分子在水平方向与竖直方向均发生了54.154°的旋转。
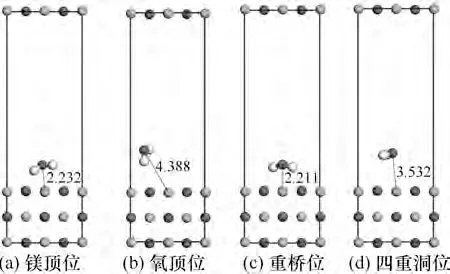
图9 H2O在MgO(001)表面4种构型下吸附后结构Fig.9 The structure of H2O adsorbed on MgO(001)surface under four configurations
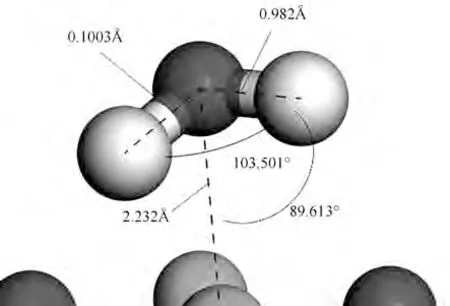
图10 镁顶位时吸附在MgO(001)表面的稳定构型Fig.10 The stable configuration of H2O adsorbed on the surface of MgO(001)at Mg-top
通过电荷布居分析可以定量讨论电荷的转移情况,本研究对以上4 种吸附结构的水分子中O 和H 原子的电荷布居进行分析,结果如表3 所示,表中数据表明,H2O 在MgO(001)表面的吸附是典型的化学吸附。在吸附成键过程中,O 原子获得电子表现为非金属性,Mg原子失去电子表现为金属性。不同吸附位置O 和H 原子间的得失电子情况不同,镁顶位、氧顶位、四重洞位和二重桥位O原子得到电子数分别为0.88、0.97、0.95、0.88,这部分电子是由Mg 原子转移出来的,说明Mg 原子在相对稳定的镁顶位和二重桥位处失去电子的能力较强,失去的电子数最多[14]。由此可知,与吸附在氧顶位和四重洞位相比,水分子在MgO(001)晶面的镁顶位和二重桥位时,存在更强的化学键,分子与表面的作用力更强,形成的吸附结构也更稳定。
图11为四种吸附构型优化后的电子密度图,其中颜色深浅代表电荷密度的大小。从图中可以看出,镁顶位和二重桥位存在电子云重叠,氧顶位和四重洞位没有电子云重叠,说明镁顶位和二重桥位水分子和氧化镁晶体之间作用力较大,形成了化学键,而氧顶位和四重洞位作用力较小,未形成化学键,这与表2 的吸附能判断化学吸附/物理吸附结果相符合。此外,镁顶位的电子密度图水分子与晶体的镁和氧均有较大重合区域,而二重桥位重合区域较小,说明镁顶位发生了大量电子转移,水分子的H 原子和氧化镁中的O 原子结合成为—OH,另一部分的—OH 和氧化镁中的Mg原子连接成键,镁顶位比二重桥位更利于电子转移,也就是说更容易形成稳定化合物,即对反应更有利,与表2、表3的结果吻合。

表3 不同吸附位置O原子与H原子的电荷布居数Table 3 The charge population of O and H atoms at different adsorption positions
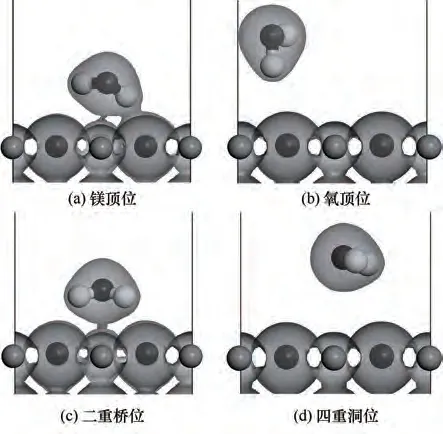
图11 四种吸附构型优化后的电子密度图Fig.11 Electron density map of four adsorption configurations after optimization
2.3 氧化镁热力学性能分析
为分析温度对氧化镁反应难易程度的影响,本研究对氧化镁的能量和温度进行模拟计算,结果如图12 所示。其中曲线①表示温度(T)与熵(S)的乘积与温度之间的关系。熵是体系混乱程度的量度。由图可知,TS 随温度升高而增大,增大趋势也在增加,即曲线斜率随温度升高而增大。原因是温度升高,氧化镁晶体内的原子热运动加剧,熵增,温度上升越快,运动越剧烈。曲线②为焓(H)与温度(T)之间的关系。焓是热力学中表征物质系统能量的一个重要状态参量,等于内能(U)与压强和体积的乘积之和。温度升高,焓值增大,曲线斜率随温度升高而缓慢增大,原因是温度升高,氧化镁晶体内原子动能加大,内能增大,且系统发生热膨胀现象,压强和体积的乘积增大,故而焓值增大。曲线③为Gibbs 自由能(G),等于焓与温度与熵乘积的差值,即曲线②与曲线①的差值。可以利用Gibbs 自由能值判断反应发生的自发性,还可以求热力学平衡常数,以判断采用升温还是降温的方法促进反应进行[32]。等温、等压的封闭体系内,不做非体积功的前提下,任何自发反应总是朝着G 减小的方向进行。当温度升高时,焓升高,熵也增加,熵与温度的乘积升值远远大于焓的升值,温度越高,其差距越大,自由能的值越低,且恒不大于0,说明能够反应的趋势随温度升高而增大[33],这对寻找适宜储热温度有很好的指导意义。
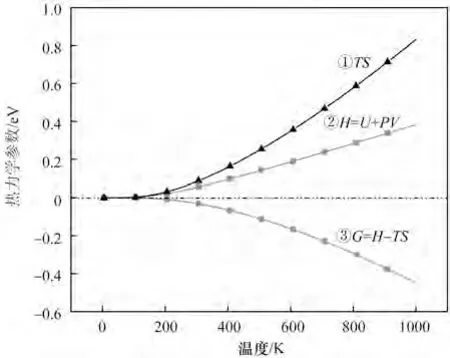
图12 热力学参数随温度变化的规律Fig.12 The variations of thermodynamic parameters with temperature
德拜温度是反应分子结合力的一个重要的指标,弹性、硬度、熔点和比热容等多个物理量都与之相关。对于比热容,当温度高于德拜温度时,固体的比热容符合Dulong-Petit 定律,Cv=3R(Cv为比定容热容);温度低于德拜温度时,比热容遵循量子规律,比热容与温度的三次方呈正比。物质的熔点越高,其原子结合力越强,德拜温度越高。
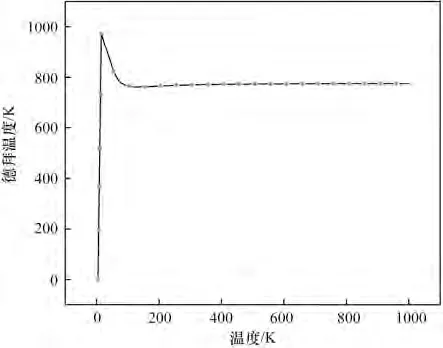
图13 德拜温度随温度变化规律Fig.13 The variation of Debye temperature with temperature
图13中,在0~15 K,随着温度升高,德拜温度由0 急剧升高到975.2 K;在15~95.45 K,德拜温度由975.2 K 降低至796.7 K;在95.45 K 往后,随着温度升高,德拜温度几乎趋于稳定,不再变化。可根据其计算出比热容。
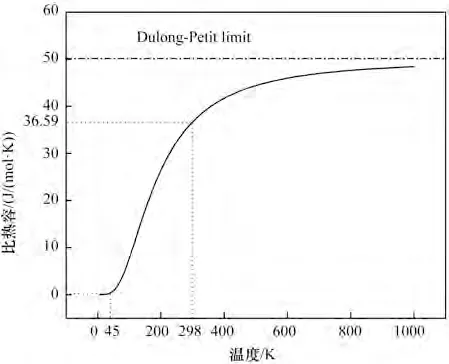
图14 MgO比热容随温度变化的规律Fig.14 The variation of heat capacity of MgO with temperature
图14展示了热容量与温度之间的关系,在极低温度时(0~45 K),比热容接近0;温度在45~764 K时,比热容随着温度升高而递增,曲线的斜率由大变小,渐渐呈现饱和趋势,最终在900 K 左右趋近于常数。由图13可知,温度在764 K时,此时温度低于德拜温度,比热容遵循德拜T3定律,与热力学温度的三次方呈正比,这是因为温度较低时,固体的热容量是由晶格热振动和电子热运动共同作用,热容量增长较快;而温度较高时,比热容趋于稳定,这是由于受非谐效应的影响,从而导致比热容几乎接近Dulong-Petit 值,这部分主要是由晶格热振动决定。在常温下得到的比热容值为36.59 J/(mol·K),这个值和逯来玉[27]得到的36.53 J/(mol·K)相一致。

图15 有/无掺杂过渡态能量对比Fig.15 Reaction energy profiles with/without doping
2.4 掺杂对吸附的影响
过渡态是反应过程中旧键断裂、新键形成的中间状态,其能量处于最高状态,此时能量与反应物的能量差(ΔE)为活化能,活化能越小,即过渡态越低,反应越容易进行。本研究对Li、Na、K 三种原子掺杂于氧化镁晶体进行过渡态对比,如图15 所示,图中超晶胞表层灰色原子(位于超晶胞表层边缘中心的两个原子,其相互轴对称,实际占据超晶胞中镁原子的1/12)为掺杂原子所在位置,计算结果发现,Li、Na、K 三种原子对反应的过渡态有降低作用。过渡态只能通过基元反应获得,从微观角度来看,过渡态的降低代表着储热条件的降低,在温度一定时,储热速度可以由过渡态的变化进行推测。在掺杂Li、Na、K 三种原子后,过渡态分别降低了0.1952、0.1104、0.0160 eV,能量势垒明显降低,也就是说反应更容易实现,反应可以在更低的温度下实现,且掺杂的金属对应的原子活泼性越高,过渡态降低幅度越大,越有利于反应的进行。
针对掺杂的不同原子的热力学性能进行了研究,如图16 所示,由图可知,掺杂以后熵增,K 对熵增的影响最大,Li 和Na 影响程度接近[图16(a)];掺杂能增加氧化镁的比热容值,Li 和K 对比热容影响较大,且二者接近,Na 对氧化镁的热容影响稍逊之[图16(b)];掺杂能降低反应的自由能,其中掺杂Li和K 的降低程度更大,Na 次之,表明掺杂更有利于反应的进行,这和图15中的结果相一致[图16(c)];掺杂对反应的焓值影响较小,即对储能密度影响较小[图16(d)]。综合来看,掺杂K 对储热促进效果最好,其次是Li,Na 最差。这是因为掺杂金属原子对晶体结构的对称性造成了破坏,使得化学键更容易断裂,反应更容易进行,新的物质更容易生成。

图16 掺杂不同种原子热力学性能对比(1 cal=4.1868 J)Fig.16 Comparison of thermodynamic properties of different doping atoms
3 结 论
本文在微观机理层面建立了相应储热材料的分子晶体结构模型,采用第一性原理密度泛函理论,分析了不同吸附距离和吸附构型对吸附稳定性的影响,从微观方面模拟掺杂了Li、Na、K 三种金属原子,研究了其对储热的影响,主要结论如下。
(1)研究了水分子和底物氧化镁不同距离对吸附能的影响,发现在物理吸附和化学吸附共同作用下水和氧化镁的最佳吸附距离为0.15 nm。
(2)分别从吸附能、原子间距离变化、键长变化、电子密度云等方面对氧化镁、水系统四种不同吸附构型进行对比分析。结果表明,氧顶位和四重洞位的构型中存在原子互斥,对成键不利,导致吸附能较小;二重桥位构型有利于新键生成,吸附效果较好;Mg顶位具有高对称性,有利于电子转移,吸附性能最好,最符合实际情况。
(3)对于氧化镁和水的反应,分别掺杂Li、Na、K三种金属原子进行研究,发现掺杂金属原子对晶体结构的对称性造成了破坏,使得化学键更容易断裂,反应更容易进行,新的物质更容易生成,所以掺杂后熵、比热容都发生了不同程度的增大,且均能一定程度地降低反应的自由能,反应更容易进行,这与已有研究的实验结果吻合较好。
本文对探索基于元素掺杂的热化学储热材料改性方法、揭示相关热化学储热微观系统的反应机理有很好的指导意义。
