基于分子动力学模拟的CH4溶解对原油分子间作用的影响机制研究
2021-04-09李秉繁刘刚陈雷
李秉繁,刘刚,陈雷
(中国石油大学(华东)山东省油气储运安全省级重点实验室,山东青岛266580)
引 言
原油生产和输送过程中必然面临着轻组分的析出和溶解,这会显著影响原油宏观流变特性,进而产生一系列生产问题[1-4]。近年来,国内外学者[5-11]围绕溶气原油的气体溶解度、倾点、黏度等参量开展了大量宏观试验研究,发现在原油达到饱和前,随着溶气压力的增大,原油溶解量逐渐增大,原油倾点、黏度及屈服应力均逐渐减小,CH4等轻组分溶解对原油具有降黏作用,并通过试验研究提出了相应气体溶解对原油降黏的作用机理,包括稀释作用机理[5-7]、溶胀作用机理[8-10]、气泡作用机理[11]等。然而,宏观试验不能得出造成以上机理的原因,阻碍了上述降黏机理的应用。
分子动力学模拟以经典牛顿力学为基础,能够分析分子间作用力、分子形态等微观性质的变化[12-25],可以作为宏观试验的补充。目前,分子动力学模拟在石油领域已得到了相应应用。学者[12-16]利用分子模拟手段开展了气体在不同石油相关材料中的吸附、溶解行为研究。Stubbs 等[12]采用Monte Carlo 方法模拟了超临界CO2+甲醇体系在结构、相平衡、氢键等方面的特性;Brochard 等[13]通过Monte Carlo 法模拟了CO2、CH4分子在煤表面的竞争吸附证明了CO2的注入使得CH4解吸附;Jin 等[14]利用Monte Carlo 法模拟研究水对黏土矿物中CH4和CO2吸附的影响,结果表明水的加入使得黏土纳米孔中的CH4和CO2吸附量显著减少;Zhang 等[15]利用分子动力学方法研究了CO2对辛烷(正辛烷)溶胀的影响以及在辛烷中的溶解度,并对两者的相容性进行了分析;Liu 等[16]利用分子动力学模拟研究了CO2对4种烷烃体积膨胀的影响,并通过分子间间距、相互作用能分析CO2对烷烃溶胀的影响机理。Zabala等[17]借助分子模拟手段获取了CO2-正构烷烃体系的扩散系数,而根据分子模拟的基本理论,扩散系数与体系黏度存在对应关系[18-19]。因此分子模拟常被用于混合体系理论黏度的分析研究。部分学者利用分子模拟进行胶质、沥青质混合体系的稳定性研究[20-25],通过分析分子间作用力探索不同组分对聚合体系稳定性的作用规律。Rogel[20]认为维持胶质、沥青质聚合体稳定性的能量主要是分子间的范德华力,且聚合体稳定能量大小取决于胶质、沥青质分子的组成和结构特性;Takanohashi 等[21-22]指出芳环-芳环、范德华力等几种非共价相互作用聚集的沥青聚合体结构最稳定;Zhang 等[23-24]利用分子动力学模拟研究沥青的密度、体积模量等物理性质和微观结构,模拟结果与实验结果高度一致;李英锋[25]利用Material Studio 分析了温度对沥青质缔合体分子结构的影响。
综上所述,分子动力学模拟在材料的溶解行为、黏度计算和稳定性等方面取得了相应研究成果。因此本文采用分子动力学方法,分别构建了正庚烷与正庚烷、蜡分子、胶质、沥青质的最低能量构型,分析CH4氛围对原油分子间相互作用的影响,并以CH4/原油分子体系模型为研究对象进行分子动力学模拟,考察CH4溶解对原油分子体系黏度的影响,根据原油分子间相互作用、径向分布函数、体积应变、自扩散系数以及体系内聚能密度变化规律,研究CH4溶解对原油分子间作用的影响机制,为原油溶气改性技术研究提供理论依据。
1 原油分子体系构建及评价
含蜡原油的主要成分为烷烃、环烷烃、芳香烃、胶质和沥青质等,鉴于原油化学组成、结构的复杂性,建立与原油组成和性质完全对应的模拟体系几乎不可能。由于实验室含蜡模拟油主要通过基础油、切片石蜡、胶质、沥青质混合配制而成[26-27],因此本文模拟选取实验室所用的基础油正庚烷(C7H16)作为溶剂油体系,20℃密度为0.6835 g/cm3;根据实验室切片石蜡的碳数平均值选取正构C24H50模拟蜡分子,20℃密度为0.7984 g/cm3;胶质是原油中分子量相对较重的组分,通常为长支链稠环化合物或极性芳香环稠环化合物,但胶质中芳香环的个数均小于7 个[28],20℃密度约为1.07 g/cm3,胶质平均分子模型选用Modified R-benzo-thio-S 分子模型[29];沥青质是原油中密度和分子量最大的组分,均高于胶质,但两者化学结构相似,20℃密度约为1.15 g/cm3,沥青质平均分子模型选用At-N+2S+O 分子模型[30]。选取的原油组成各分子模型如图1所示。

图1 原油组分分子模型Fig.1 Molecular model of crude oil components
根据文中选用的正庚烷、正构C24H50、胶质平均分子模型、沥青质平均分子模型,利用Materials Studio 中Visualize 模块分别建立相应的分子模型,并对每一个分子模型进行几何优化以及能量最小优化得到原油分子模型的最优化结构构型,同时构建CH4分子模型。表1 为常压条件下胜利原油和南阳原油基本物性,根据表1 中两种原油蜡、胶质、沥青质的质量分数换算为相应的摩尔分数,如表2所示。

表1 常压条件下油样基本物性Table1 Basic physical properties of oil samples under atmospheric pressure

表2 周期性体系中原油分子数量Table 2 The number of molecular in periodic systems
利用Amorphous Cell Tools 模块下Construction工具构建两种原油分子体系,如图2所示。

图2 原油分子体系(灰色代表正庚烷、红色代表蜡分子、蓝色代表胶质分子、绿色代表沥青质分子、黑色为氢原子)Fig.2 System of crude oil molecular
密度是原油基础物性之一,是评价所建立的原油分子体系合理性的重要依据。利用Forcite 模块中Dynamics 对原油分子体系进行密度计算,通过对比模拟密度值与试验参考密度值,验证模拟体系的合理性。动力学模拟参数为:NPT 系综,CPMPASS力场,温度为293.15 K,压力为常压,静电交互和范德华交互分别采用Ewald 和Atom Based 方法,截至距离1.25 nm,Anderson 法控温,Berendsen 法控压,步长为1fs 进行100 ps(100000 步)动力学模拟,每500步输出一次运行结果。
利用分子动力学模拟计算常压20℃条件下原油分子体系密度,如图3 所示。分子动力学模拟计算的时间为100 ps,其中前40 ps 用于平衡原油分子体系,后60 ps用于模型密度数据采集。利用文献[31]中所述的方法进行密度平衡判定,即当标准差小于0.001 时,认为体系达到平衡状态。由图3 和表3 可知,原油分子体系的密度随模拟时间延长逐渐趋于平衡(标准差均小于0.001),对40~100 ps 时间范围内原油分子体系密度求平均值,并与试验参考密度值进行对比,如表3所示。
由表3 可知,分子动力学模拟计算的原油分子体系密度值接近试验参考密度值,且绝对误差均小于3.773%,表明所建立的原油分子体系以及模拟流程的准确性。
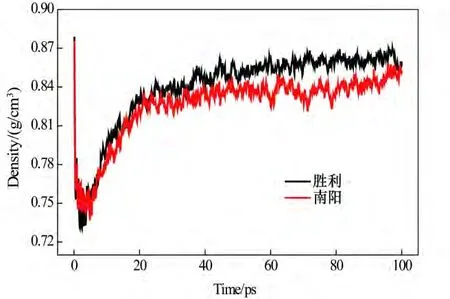
图3 原油分子体系密度随模拟时间变化关系Fig.3 The relationship between the density of crude oil molecular system and simulation time
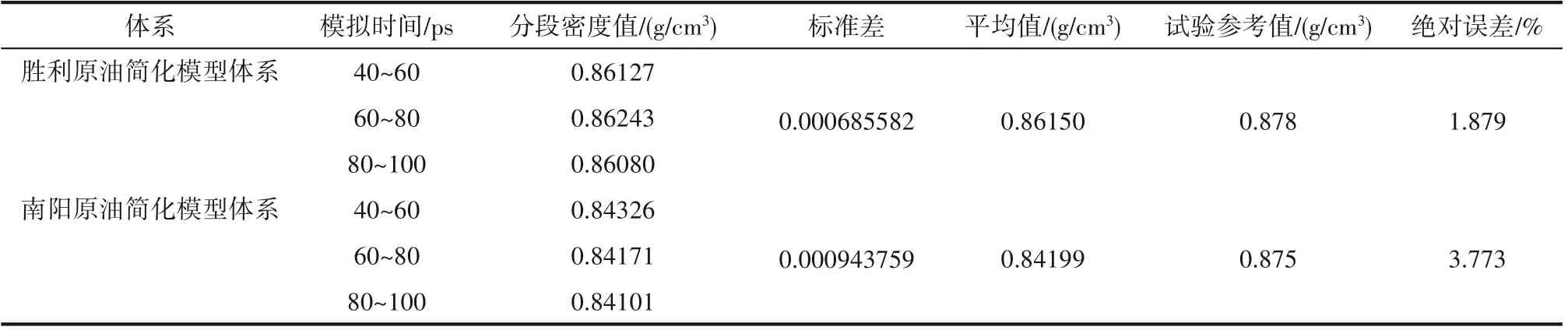
表3 模拟密度与试验参考密度对比Table 3 Comparison between simulated density and test reference density
为了获得CH4/原油分子体系,利用Sorption 模块对CH4在不同原油分子体系中的溶解进行巨正则Monte Carlo 计算。计算过程采用周期性边界条件;任 务 选 择 Adsorption isothem;平 衡 步 数(equilibration steps) 为 2000000;产 出 步 数(production steps)为1000000;力场为COMPASS;静电交互和范德华交互分别采用Ewald 和Atom Based方法;溶质为CH4;压力为常压到20 MPa,温度为20℃。
图4为16 MPa压力条件下CH4在胜利原油简化模型体系和南阳原油简化模型体系中溶解的构型图。为了观察清晰,删除了氢原子,蓝色区域为体系的自由体积空隙,蓝色区域越大,自由体积越大。图5 为温度为20℃时不同压力条件下CH4在原油分子体系中溶解过程的构型图。图4、图5中黄色球状为CH4分子、链状为原油分子。

图4 16 MPa条件下CH4在原油分子体系中溶解的构型图Fig.4 Configuration diagram of CH4 dissolution in crude oil molecular system at 16 MPa
由图4 可知,不同分子组成的原油分子体系中存在较多的空隙,溶解的气体主要集中在这些空隙中。Everett 等[32]通过计算狭缝孔中吸附原子的势能分布发现,随着空隙直径的减小,固体表面与吸附质分子相互作用势增大。CH4要在原油体系中溶解,必须克服空隙壁产生的势垒。当溶气压力较小时,原油分子会吸引CH4使其溶解到原油体系空隙的高能吸附位,CH4溶解位置多为空隙较大的地方;随着溶气压力的增大,CH4能够克服更高的势垒进入空隙较小的地方,即随着溶气压力的增大CH4的溶解量逐渐增多。如图5 所示,随着溶气压力的增大,CH4溶解量逐渐增大,但溶解的位置可能发生变化,这是由于溶解的CH4分子仍处于振动状态,若通过吸热获得一定能量,分子的布朗运动增大,就可能摆脱碳原子的引力作用重新成为自由气体;当溶气压力较低时,CH4分子较为分散地分布在原油体系中;随着溶气压力的增大,CH4的溶解量逐渐增多,部分溶解的CH4分子会发生聚集,如图4 所示。

图5 不同压力条件下CH4在原油分子体系中溶解过程的构型图Fig.5 Configuration diagram of CH4 dissolution in crude oil molecular system at different pressure
图6 为不同压力条件下CH4分子在原油体系中溶解量曲线。随着压力增大,溶解量先增大后逐渐趋于平衡,CH4的溶解符合孔填充机制[33-34]。随着溶气压力的增大,CH4在原油分子体系中不断填充,直至体系空隙填充满为止,到达最大饱和溶气压力后,气体溶解量基本不变,胜利原油简化模型体系和南阳原油简化模型体系最大饱和溶气压力分别为11.44、13.89 MPa。

图6 不同压力条件下CH4分子在原油分子体系中溶解量曲线Fig.6 Solubility curve of CH4 in crude oil under different pressure
2 CH4溶解对原油分子体系黏度的影响规律
利用Forcite Plus 模块下的Dynamics 工具分别对CH4/原油分子体系模型(图5)进行分子动力学模拟。动力学模拟参数为:NPT 系综,Compass 力场,温度为20℃,压力为常压到20 MPa,静电交互和范德华交互分别采用Ewald 和Atom Based 方法,截至距离1.25 nm,Anderson 法控温,Berendsen 法控压,步长为1 fs 进行100 ps(100000 步)动力学模拟,每5000步输出一次运行结果。
分子动力学计算流体黏度方法包括基于Couette 或Poiseuille 流动的非平衡分子动力学系统黏度计算方法和基于原子平衡态条件下的运动状态流体黏度计算方法[35]。基于平衡系统分子动力学系统计算流体黏度常用的方法有两种,包括Stokes-Einstein 关系和Green-Kubo 应力相关函数黏度计算方法,本文利用Green-Kubo 应力相关函数关系计算原油分子体系黏度。
式(1)为Green-Kubo关系[35]:

基于分子动力学方法对20℃不同溶气压力条件下胜利原油简化模型体系和南阳原油简化模型体系零剪切黏度进行模拟,模拟结果如图7所示。
由图7 可知,随着溶气压力的增大,由于CH4溶解降黏作用使得原油分子体系黏度逐渐降低,但降低的幅度逐渐减小,在溶气压力达到最大饱和溶气压力时,溶气压力的增黏效果与CH4溶解的降黏效果相同,CH4溶解降黏效果达到最佳,随着溶气压力的进一步增大,溶气压力的增黏效果起主导作用,原油分子体系黏度逐渐增大。

图7 原油分子体系零剪切黏度与溶气压力关系曲线Fig.7 Relationship between zero shear viscosity and dissolved gas pressure of crude oil molecular system
3 CH4溶解对原油分子间作用的影响机制
结合CH4溶解对原油分子体系黏度的影响,从CH4氛围对原油分子间相互作用的影响、径向分布函数、体积应变、自扩散系数和体系内聚能密度5个方面开展CH4溶解对原油分子间作用的影响机制研究。
3.1 CH4氛围对原油分子间相互作用的影响
原油是一种较为稳定且复杂的胶体分散体系,其中分散相以沥青质、胶质为核心,分散介质为液态油组分。对于含蜡原油,高温条件下蜡分子基本能够溶解于原油中;当温度降低时,原油中溶解的蜡分子达到饱和并逐渐析出,蜡晶成为主要分散相。因此本文分别构建了正庚烷与正庚烷、蜡分子、胶质、沥青质的最低能量构型,如图8(a)、(c)、(e)、(g)所示。
为了研究CH4溶解对原油分子间相互作用的影响,将1000 个CH4分子尽量均匀地放置在4 种最低能量构型体系的周围,如图8(b)、(d)、(f)、(h)所示(删除了CH4分子)。其中,模拟采用正则系综,Compass力场。
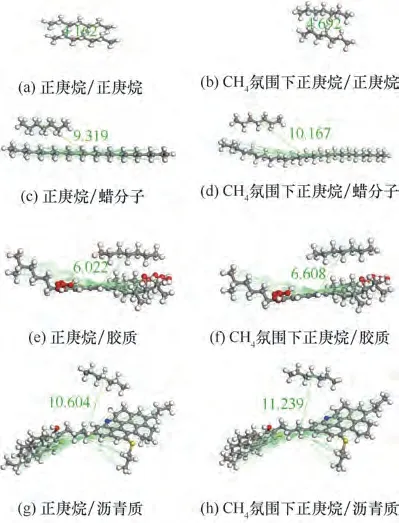
图8 20℃条件下不同分子间的最低能量构型和CH4氛围下不同分子间的最低能量构型Fig.8 The lowest energy configuration of different molecules and that of different molecules in CH4atmosphere at 20℃
表4 为CH4对正庚烷与正庚烷、蜡分子、胶质、沥青质分子间间距及相互作用能的影响对比。由表4、图8 可知,无论是否在CH4氛围下,不同分子间间距和分子间作用能大小依次为:正庚烷/正庚烷>正庚烷/胶质>正庚烷/蜡分子>正庚烷/沥青质。这是由于胶质分子存在碳数较少的支链,胶质的多臂结构可以提供更多的与正庚烷的结合位点,使得胶质与正庚烷的相互作用增强;而正庚烷是碳数为7 的长直链烷烃,蜡分子是碳数为24 的长直链烷烃,沥青质是碳数为59 且多为芳香环的大分子,根据相似相溶原理,分子结构相似,分子越容易聚集在一起,相互作用强度越大,说明碳数是影响分子相互作用的重要因素,但不是单一决定因素,还与支链相关。
随着CH4的加入,正庚烷/正庚烷、正庚烷/蜡分子、正庚烷/胶质、正庚烷/沥青质分子间间距均增大,且分子间作用能均减小。这是由于在CH4氛围条件下,原油分子间的范德华引力逐步转化为原油分子与CH4分子间的作用,体系内平均分子间间距增大、平均分子间相互作用能减小。为了分析CH4对不同分子间间距以及分子间作用能的影响程度,分别计算分子间间距的相对改变量和分子间相互作用能的相对改变量,计算结果如表4 所示。可以看出CH4的加入,对不同分子间间距影响程度大小依次为:正庚烷/正庚烷>正庚烷/胶质>正庚烷/蜡分子>正庚烷/沥青质,对分子间作用能影响程度大小依次为:正庚烷/正庚烷>正庚烷/蜡分子>正庚烷/胶质>正庚烷/沥青质,表明CH4的溶解主要通过增大正庚烷/正庚烷分子间距增大原油体系体积,且影响正庚烷/蜡分子的相互作用改善油品的流变性。
3.2 径向分布函数
径向分布函数(RDF)是指体系在参考粒子处区域面积与体系平均密度之比,能够反映体系分子聚集程度大小以及体系内部的微观结构。在模拟过程中原油分子是不断变化的,通常采用体系的一些特征性质(如回转半径、径向分布函数)进行统计平均描述分子结构,而RDF 通过傅里叶变换与结构因数相关联[36]。因此本文采用RDF描述原油分子体系的结构相关性和有序性。
图9为不同溶气压力条件下胜利原油简化模型体系和南阳原油简化模型体系内C—C 原子间径向分布函数。由图可知,不同原油分子体系的径向分布函数变化趋势基本相同,且在0.6 nm 以内径向分布函数出现不同程度的函数峰值,表明原油分子体系出现不同程度的分子聚集,其中在0.153 nm 处RDF峰值最大,碳原子聚集最多;0.6 nm以外径向分布函数逐渐平缓趋近于1,表明不同溶气压力条件下原油分子体系分子呈现近程有序、远程无序状态。通过对比不同溶气压力条件下原油分子体系径向分布函数发现,在最大饱和溶气压力范围内,不同原油分子体系的径向分布函数峰值随着溶气压力的增大而减小,表明随着溶气压力的增大,原油分子体系溶解的CH4气体逐渐增多,使得体系内原油分子的聚集程度逐渐降低,能够有效减小原油分子的靠近、阻碍分子链聚集成团,减少了原油分子间的纠缠,进而改善原油流变性。当溶气压力超过最大饱和溶气压力时,随着溶气压力的增大原油分子体系的径向分布函数峰值逐渐增大,使得体系内原油分子的聚集程度逐渐提高。通过对比相同溶气压力条件下原油分子体系的径向分布函数发现,CH4溶解对不同原油分子体系径向分布函数影响变化趋势类似,原油大分子的存在影响原油分子体系的径向分布函数大小,但未改变CH4对原油分子体系微观结构的影响机制。

表4 正庚烷与正庚烷、蜡分子、胶质、沥青质分子间间距及相互作用能Table 4 Intermolecular spacing and interaction energy of n-heptane with n-heptane,wax,colloid and asphaltene

图9 C-C原子间径向分布函数Fig.9 Radial distribution function between C-C atoms
3.3 体积应变
图10 为不同原油分子体系的体积应变。随着溶气压力的增大,CH4分子逐渐溶解,不同原油分子体系的体积应变均逐渐增大,但增大趋势逐渐减缓,当达到最大饱和溶气压力,随着溶气压力的进一步增大,体系体积应变逐渐减小。原油分子体系体积应变由气体溶解引起的膨胀和溶气压力的压缩作用共同影响,在最大饱和溶气压力范围内,随着溶气压力的增大原油分子体系体积应变逐渐增大,气体的溶解膨胀起主导作用;当溶气压力达到最大饱和溶气压力时,气体溶解引起的膨胀和溶气压力的压缩作用效果达到平衡,原油分子体系体积应变达到最大;随着溶气压力的进一步增大,原油分子体系体积应变逐渐减小,溶气压力的压缩作用起主导作用。
随着溶气压力的增大,溶解的CH4与原油分子通过分子间作用力而形成有机的整体,从而促进体系的体积膨胀,CH4的溶解增大了原油分子间的间距,削弱了体系内原油分子间的作用强度;体积膨胀为原油分子的跳跃和迁移提供更广的空间,分子热运动加剧,体系内动能增加,油品流动过程中流动阻力和毛细管阻力减小,使得油品的流动能力增强。随着溶气压力的进一步增大,体系体积应变逐渐减小,减少了原油分子的跳跃和迁移的空间,使得油品流动性变差,黏度增加。
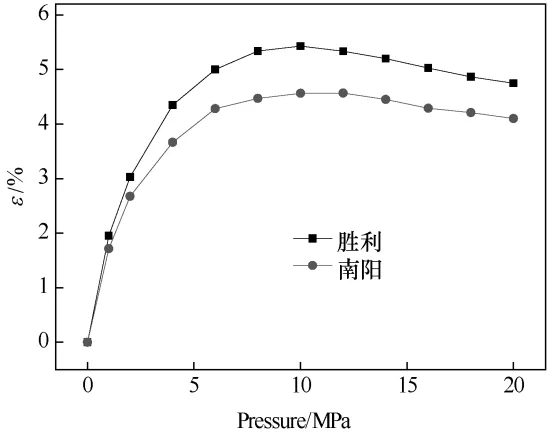
图10 不同原油分子体系的体积应变Fig.10 Volume strain of crude oil molecular systems
3.4 原油分子的自扩散
自扩散是通过体系内粒子扩散轨迹的均方位移(MSD)计算,可以描述宏观平衡状态下分子的运动情况,能够反映原油分子运动的剧烈程度。原油分子的自扩散系数D可以由Einstein关系式计算[37]:
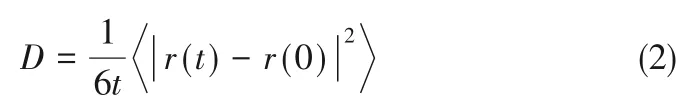
式中,|r(t) - r(0)|2为原油分子质心的均方位移,即:

由于均方位移(MSD)与时间关系曲线的斜率a可近似代替式(2)中的微分,且MSD 的值已经对分子个数N 做了平均[38],因此Einstein 关系式可简化为:


图11 20℃条件下不同溶气压力条件下原油分子的均方位移Fig.11 Mean square displacement of crude oil molecules under different dissolved gas pressures at 20℃
图11 为20℃时不同溶气压力条件下原油分子的均方位移。不同原油分子体系的原油分子均方位移与模拟时间基本呈线性关系,在最大饱和溶气压力范围内,原油分子均方位移逐渐增大,说明原油分子的扩散迁移能力逐渐增强;当溶气压力超过最大饱和溶气压力时,随着溶气压力的增大原油分子均方位移逐渐减小,原油分子的扩散迁移能力逐渐减弱。对于不同溶气压力条件下的原油体系,一方面CH4溶解增大原油分子的均方位移,CH4气体的溶解,使得原油分子间间距逐渐增大,分子间作用力逐渐减小,原油分子运动逐渐加剧;另一方面溶气压力削弱原油分子的均方位移,原油分子均方位移变化是这两个因素综合影响的结果。通过对比不同溶气压力条件下不同原油分子体系的自扩散系数发现,CH4溶解对原油分子自扩散的影响变化趋势类似,原油大分子的存在影响原油分子自扩散系数的大小,但未改变CH4对原油分子跃迁和驱替规律。
通过式(4)计算得到20℃条件下不同溶气压力条件下原油分子的自扩散系数,如表5 所示。不同原油分子体系的自扩散系数变化趋势类似,在达到最大饱和溶气压力之前,原油分子的自扩散系数随着溶气压力的增大而增大,原油分子的自扩散系数的增大能够较为容易地翻越能量势垒实现分子的跳跃迁移,流体流动性能提高;在达到最大饱和溶气压力之后,原油分子的自扩散系数随着溶气压力的增大而减小,原油分子运动能力减弱,流体流动性能变差。
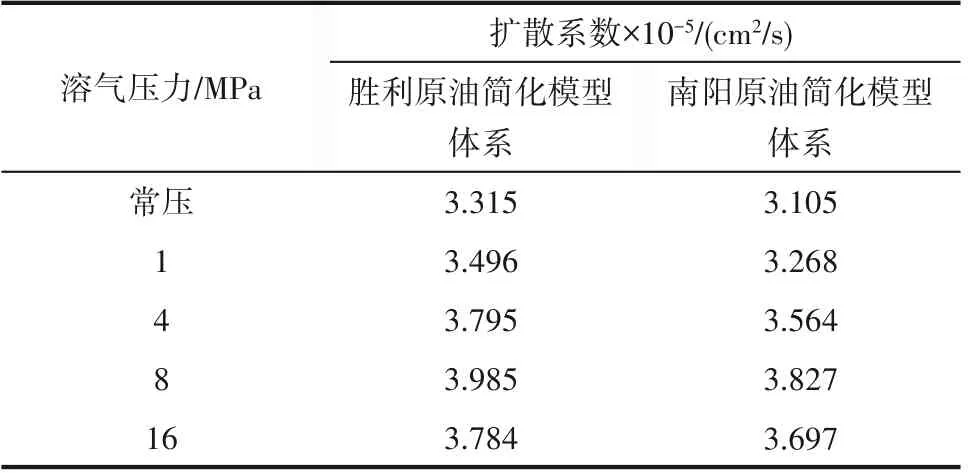
表5 不同溶气压力条件下原油分子的扩散系数Table5 Diffusion coefficient of crude oil molecules under different dissolved gas pressures
根据Stokes-Einstein 关系式[39][式(5)]可知,原油分子体系黏度和自扩散系数呈反比关系,原油分子体系自扩散系数越小,黏度越大。在最大饱和溶气压力范围内,随着溶气压力的增大不同原油体系自扩散系数均逐渐增大,黏度逐渐减小,油品流动性得到改善;当溶气压力超过最大饱和溶气压力时,随着溶气压力的增大不同原油体系自扩散系数均逐渐减小,油品黏度增加。

3.5 体系内聚能密度
体系的内聚能(cohesive energy)是指将体系内的所有分子相互分开至无限远所需的平均能量,而内聚能密度(cohesive energy density,CED)是指单位体积的内聚能,是评价分子间作用力大小的物理量[40-41]。在MS 模拟Compass 力场中,CED 实际上被视作克服分子间的非键力所需的能量,即为克服范德华力和静电力所需的能量之和。
表6为利用分子动力学模拟计算得到不同溶气压力条件下原油分子体系的内聚能密度及分量。在最大饱和溶气压力范围内,随着溶气压力的增大两种原油分子体系的内聚能密度均逐渐减小,CH4的溶解改变了原油体系内分子聚集状态,降低了体系内聚能密度;相比于溶气原油体系,常压条件下原油体系内聚能密度更大,若两类体系达到相同流动状态,常压条件下原油体系需外界提供更多的能量。当溶气压力超过最大饱和溶气压力时,随着溶气压力的增大两种原油分子体系内聚能密度均逐渐增大,溶气压力对原油体系内分子聚集状态影响程度起主导作用,增大了体系内聚能密度,流体流动需要外界提供更多的能量。

表6 不同溶气压力条件下原油分子体系的内聚能密度及分量Table 6 Cohesive energy density and component of crude oil molecular system under different dissolved gas pressures
由表6 可知,静电力内聚能密度分量基本趋于零,说明原油及溶气原油体系内的库仑作用非常小,基本可以忽略不计;在达到最大饱和溶气压力前,范德华力内聚能密度分量随着溶气压力的增大而减小,且范德华力内聚能密度分量远大于静电力内聚能密度分量,这说明对于原油及溶气原油体系的流动主要克服分子间范德华作用。通过对比相同溶气压力条件下原油分子体系的内聚能密度发现,CH4溶解对不同原油体系内聚能密度影响变化趋势类似,原油大分子的存在只影响原油体系的内聚能密度大小。
4 静压力对原油分子体系黏度的影响机制
为了从分子角度解释静压力对原油分子体系黏度的影响,利用分子动力学模拟计算20℃时不同静压力条件下胜利原油简化模型体系和南阳原油简化模型体系的零剪切黏度、体积应变、自扩散系数和内聚能密度。
由表7可知,随着静压力的增大,两种原油分子体系的零剪切黏度逐渐增大、体系体积应变逐渐减小、自扩散系数逐渐减小、体系内聚能密度逐渐增大。表明随着静压力的增大,原油分子的运动剧烈程度减弱,原油分子链之间的纠缠增多,原油体系的内摩擦增大;原油分子间的间距减小,增大了体系内原油分子间的作用强度,原油分子热运动空间减小;相比于常压原油体系,高压原油体系结构更加稳定,若两类体系达到相同流动状态,高压原油分子体系需外界提供更多的能量。
5 结 论
为了研究CH4溶解对原油分子间作用的影响机制,分别构建了正庚烷与正庚烷、蜡分子、胶质、沥青质的最低能量构型,分析CH4氛围对原油分子间相互作用的影响,并以CH4/原油分子体系模型为研究对象进行分子动力学模拟,分析不同溶气压力条件下CH4对原油分子体系微观结构的影响、体系体积应变、原油分子自扩散系数以及内聚能密度的变化规律,得到以下结论。
(1)正庚烷与正庚烷、蜡分子、胶质、沥青质分子间间距及相互作用能大小依次为:正庚烷/正庚烷>正庚烷/胶质>正庚烷/蜡分子>正庚烷/沥青质,碳数是影响分子相互作用的重要因素,但不是单一决定因素,还与支链相关;随着CH4的加入,正庚烷/正庚烷、正庚烷/蜡分子、正庚烷/胶质、正庚烷/沥青质分子间间距均增大,且分子间作用能均减小。CH4的溶解主要通过增大正庚烷/正庚烷分子间间距增大原油体系体积且影响正庚烷/正庚烷的相互作用改善油品的流变性。

表7 原油模型体系参数Table 7 Crude oil system parameters
(2)随着CH4的溶解不同原油分子体系的径向分布函数峰值逐渐减小,体系内原油分子的聚集程度逐渐降低,能够有效减小原油分子的靠近、阻碍分子链聚集成团,减少了原油分子间的纠缠,进而改善原油流变性。溶解的CH4与原油分子通过分子间作用力而形成有机的整体,从而促进体系的体积膨胀,增大了原油分子间的间距,削弱了体系内原油分子间的范德华作用;体积膨胀为原油分子的热运动提供了更多的空间,原油分子热运动加剧,使得油品的流动能力增强。
(3)溶气原油流动性受CH4溶解和溶气压力共同影响,一方面CH4的溶解逐渐破坏分子缠结结构,使得原油分子伸展程度降低,溶解的CH4与原油分子通过分子间作用力而形成有机的整体,从而促进体系的体积膨胀,增大了原油分子间的间距,削弱了体系内原油分子间的范德华作用;体积膨胀为原油分子的热运动提供了更多的空间,原油分子热运动加剧,使得油品的流动能力增强;另一方面,溶气压力压缩体系体积使得原油分子聚集、原油分子间间距减小、范德华作用增大,分子热运动减弱,油品流动性减弱。
(4)不同原油分子体系的黏度、径向分布函数、自扩散系数、体积应变以及内聚能密度的变化趋势类似,原油分子体系中蜡、胶质、沥青质的存在并未改变CH4对原油分子间作用的影响机制。
符 号 说 明
D——自扩散系数,cm2/s
kB——Boltzmann常数,J/K
N——分子个数
Pxy——压力张量的分量,Pa
R——原油分子直径,nm
ri(t),ri(0)——分别为t、t0时刻第i个分子的位置向量
T——体系温度,K
t——时间,ps
V——体系体积,nm3
η——原油分子体系黏度,mPa·s
下角标
i——第i个分子
x,y——系统方向
