尿毒营养合剂质量标准研究*
2021-04-08李正胜朴春梅周训蓉
杨 亮,曾 真,李正胜,朴春梅,周训蓉
(贵州中医药大学第二附属医院,贵州 贵阳 550003)
全球慢性肾脏病(CKD)患者数量呈增长趋势,已成为威胁人类健康的重要疾病之一[1]。CKD 导致的高磷血症和一些慢性磷酸钙稳态紊乱,统称CKD 相关矿物质及骨代谢紊乱(CKD-MBD),是CKD 的主要并发症,严重影响血液透析患者的预后[2-3]。目前,西医的诊疗方案不能完全改善CKD-MBD 状态[4]。尿毒营养合剂为我院医院制剂,根据CKD-MBD 患者脾肾亏虚、浊毒内蕴、血络瘀阻的病机特点,以黄芪、白术、党参、山药益气健脾,茯苓、薏苡仁健脾利湿,地锦草、大黄泻火解毒,建曲消食和胃,肉苁蓉、黄精补肾填精,纵观全方,既补后天之脾气,亦补先天之肾精,化瘀泄浊,补泻兼施,使脾气健运,胃复和降,肾精充盈,诸证自除。前期临床研究表明,该制剂可改善维持性血液透析患者矿物质和骨代谢状况及中医临床症状积分[5]。本研究中采用薄层色谱(TLC)法定性鉴别黄芪、茯苓、白术和山药,采用高效液相色谱(HPLC)法测定君药黄芪主要成分黄芪甲苷的含量[6],为尿毒营养合剂质量标准的建立提供依据。现报道如下。
1 仪器与试药
1.1 仪器
Agilent1260 Infinity Ⅱ型高效液相色谱仪,包括蒸发光散射检测器(ELSD),均购自美国 Agilent 公司;MS105DU 型电子天平(瑞士 Mettler Toledo 公司);KQ-500DA 型超声波清洗器(江苏省昆山市超声仪器有限公司);HH-4 型恒温水浴锅(江苏省常州澳华仪器有限公司);TG-18 型高速离心机(山东博科生物有限公司)。
1.2 试药
尿毒营养合剂(批号分别为 160501,160502,160503),缺黄芪、茯苓、白术、山药的阴性样品,均由医院制剂室提供;黄芪甲苷对照品(批号为110781-201616,含量为97.4%),黄芪对照药材(批号为 120974-201612),茯苓对照药材(批号为121117-201502),白术对照药材(批号为120925-201610),山药对照药材(批号为121137-201504),均购自中国食品药品检定研究院;硅胶G 薄层板(青岛海洋化工厂、浙江台州黄岩区路桥生化塑料厂);硅胶HSG 薄层板(烟台江友硅胶开发有限公司);乙腈、甲醇为色谱纯,其余试剂均为分析纯,水为纯化水。
2 方法与结果
2.1 定性鉴别
2.1.1 黄芪
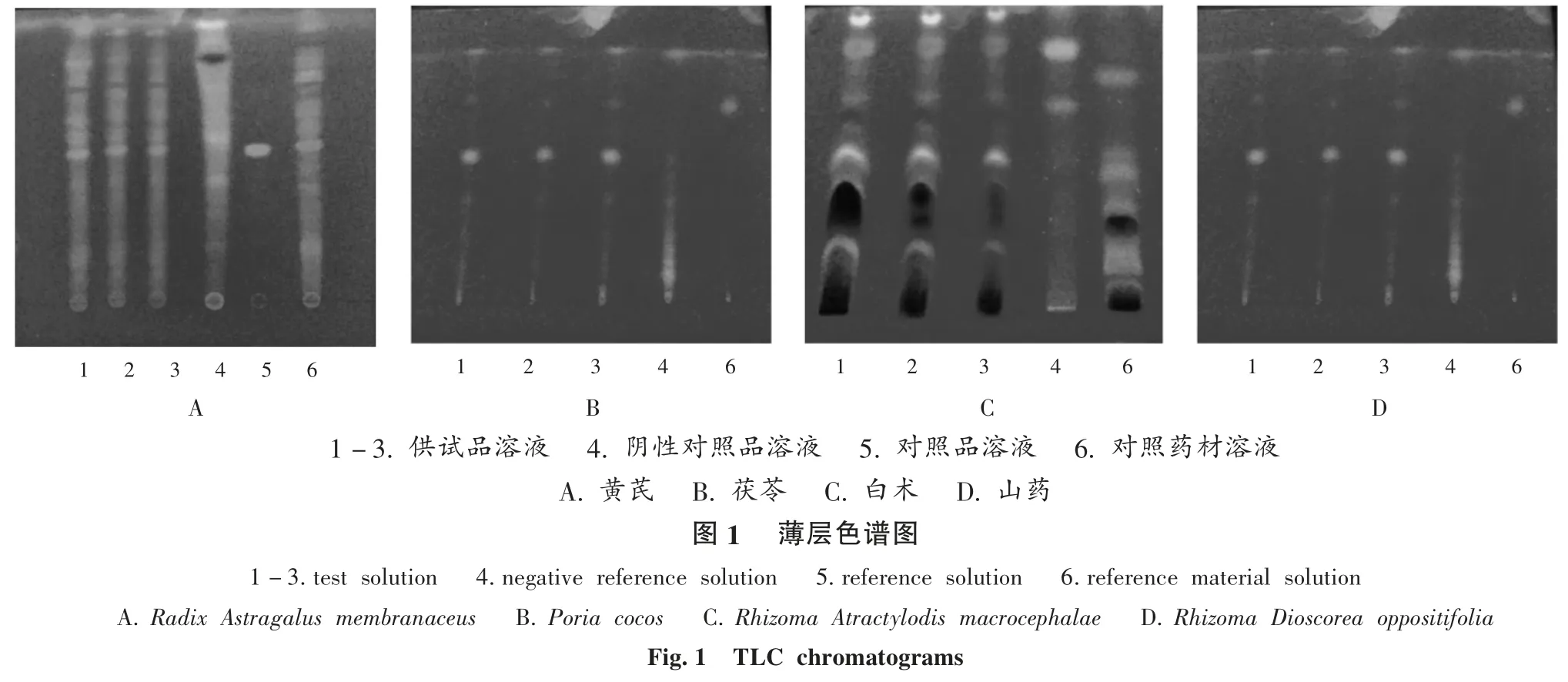
取样品适量,12 000 r/min 离心 5 min,取上清液约25 mL,加水20 mL,混匀,用正己烷振摇提取2 次,每次40 mL,弃去正己烷液,合并水液;用水饱和的正丁醇振摇提取3 次,每次40 mL,合并水饱和的正丁醇液;用氨试液洗涤2 次,每次50 mL,合并水饱和的正丁醇液;用水洗涤2 次,每次50 mL,合并正丁醇液,蒸干,残渣加甲醇1 mL 使溶解,作为供试品溶液[7]。取黄芪甲苷对照品适量,加甲醇制成每1 mL 含1 mg 的对照品溶液。取黄芪对照药材粉末2 g,精密称定,同法制备对照药材溶液。取缺黄芪的阴性样品适量,同法制备阴性对照品溶液。按2015 年版《中国药典(四部)》通则0502 项下方法试验[8],分别量取上述 4 种溶液各 10 μL,点于同一硅胶G 薄层板上,以三氯甲烷 -甲醇 -水(13 ∶7 ∶2,V / V / V)5 ~10 ℃条件下放置12 h 的下层溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,105 ℃加热至斑点显色清晰,置紫外光灯(365 nm)下检视。结果供试品溶液色谱中,在与对照品溶液及对照药材溶液色谱相应位置上显相同颜色的斑点,阴性对照无干扰[9-10],详见图1 A。
2.1.2 茯苓
取样品适量,12 000 r/min 离心 5 min,取上清液约25 mL,加水20 mL,混匀,用正己烷振摇提取2 次,每次40 mL,合并正己烷液,挥干,残渣加正己烷1 mL 使溶解,作为供试品溶液。取茯苓对照药材1 g,精密称定,加水适量,煎煮30 min,放冷,滤过,同法制备对照药材溶液。取缺茯苓的阴性样品适量,同法制备阴性对照品溶液[11]。按 2015 年版《中国药典(四部)》通则 0502 项下方法试验,分别量取上述3 种溶液各20 μL,点于同一硅胶G 薄层板上,以石油醚(60 ~ 90 ℃ )- 乙醚(1 ∶1,V / V)为展开剂,展开,取出,晾干,置紫外光灯(365 nm)下检视。结果供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同颜色的斑点,阴性对照无干扰[9-10],详见图1 B。
2.1.3 白术
取样品适量,12 000 r/min 离心 5 min,取上清液约25 mL,用乙酸乙酯振摇提取2 次,每次40 mL,合并乙酸乙酯液,挥干,残渣加乙酸乙酯1 mL 使溶解,作为供试品溶液。取白术对照药材1 g,精密称定,加水适量,煎煮30 min,放冷,滤过,同法制备对照药材溶液。取缺白术的阴性样品适量,同法制备阴性对照品溶液。按2015 年版《中国药典(四部)》通则 0502 项下方法试验[7],分别量取上述3 种溶液各10 μL,点于同一硅胶G 薄层板上,以三氯甲烷 - 乙酸乙酯(9 ∶1,V / V)为展开剂[12],展开,取出,晾干,喷以10%硫酸乙醇溶液,加热至斑点显色清晰,置紫外光灯(365 nm)下检视。结果供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同颜色的斑点,阴性对照无干扰[9-10],详见图 1 C。
2.1.4 山药
取样品适量,12 000 r/min 离心 5 min,取上清液约25 mL,用三氯甲烷振摇提取2 次,每次40 mL,合并三氯甲烷液,挥干,残渣加三氯甲烷1 mL 使溶解,作为供试品溶液。另取山药对照药材1 g,精密称定,加水适量煎煮30 min,放冷,滤过,同法制备对照药材溶液。取缺山药的阴性样品适量,同法制备阴性对照品溶液。按2015 年版《中国药典(四部)》通则 0502 项下方法试验[7],分别量取上述3 种溶液各10μL,点于同一硅胶G 薄层板上,以三氯甲烷 -乙酸乙酯(12 ∶1,V / V)为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,加热至斑点显色清晰[13]。结果供试品溶液色谱中,在与对照药材溶液色谱相应位置上显相同颜色的斑点,阴性对照无干扰,详见图1 D。
2.2 含量测定[8]
2.2.1 色谱条件
色谱柱:Waters Symmetry C18柱(150 mm ×4.6 mm,5 μm);流动相:乙腈 -水(34 ∶66,V / V);流速:1 mL /min;检测器:ELSD;柱温:30 ℃ ;漂移管温度:60 ℃[8]。
2.2.2 溶液制备
取黄芪甲苷对照品适量,精密称定,加甲醇制成每1 mL 含黄芪甲苷0.8 mg 的溶液,即得对照品溶液。取2.1.1 项下供试品溶液,置5 mL 容量瓶中,加甲醇定容,摇匀,即得供试品溶液[14]。按尿毒营养合剂处方及工艺制备缺黄芪的阴性样品,同法制备阴性对照品溶液。
2.2.3 方法学考察
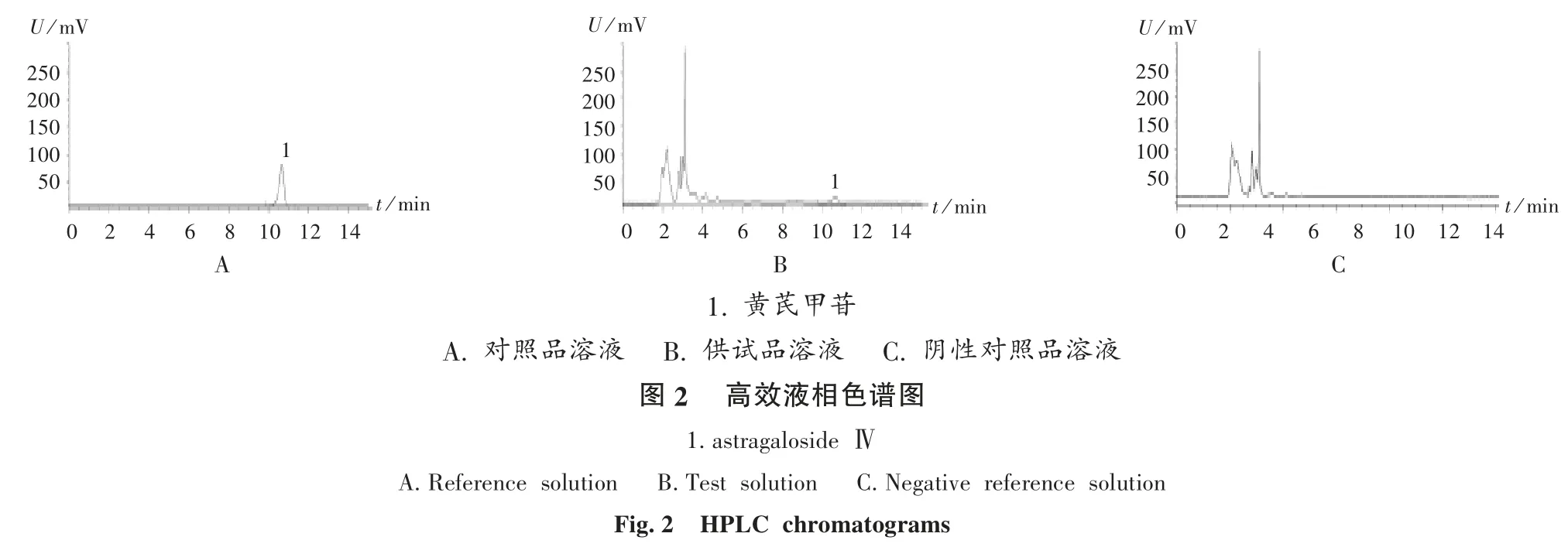
系统适用性试验:取2.2.2 项下对照品溶液、供试品溶液各适量,按2.2.1 项下色谱条件进样测定,色谱图见图2。结果理论板数按黄芪甲苷峰计应不低于3 000,分离度均大于4 000。
专属性试验:取2.2.2 项下对照品溶液及阴性对照品溶液各适量,按2.2.1 项下色谱条件进样测定,色谱图见图2。结果阴性对照品溶液色谱图中,在与黄芪甲苷对照品溶液色谱相同保留时间处无干扰峰,表明专属性良好。
线性关系考察:取黄芪甲苷对照品适量,精密称定,加甲醇制成质量浓度为0.865 3 mg/mL 的对照品溶液,精密量取 1,2,5,10,20 μL,按 2.2.1 项下色谱条件进样测定,记录峰面积。以黄芪甲苷进样量的自然对数值(X)为横坐标、峰面积自然对数值(Y)为纵坐标进行线性回归,得回归方程 Y =1.5413X-3.7802(r =0.9950)。结果表明,黄芪甲苷进样量在 0.87 ~ 17.31 μg 范围内的自然对数值与峰面积自然对数值线性关系良好。
精密度试验:取 2.2.2 项下对照品溶液适量,按2.2.1 项下色谱条件连续进样测定6 次,记录峰面积。结果黄芪甲苷峰面积自然对数值的 RSD 为0.23%(n =6),表明仪器精密度良好。
稳定性试验:取2.2.2 项下供试品溶液适量,分别于室温下放置 0,1,4,8,13,20,40 h 时按 2.2.1 项下色谱条件进样测定,记录峰面积。结果黄芪甲苷峰面积的RSD 为 0.84% (n = 7),表明供试品溶液在室温放置40 h 内基本稳定。
重复性试验:取样品(批号为160501)适量,精密称定,共 6 份,按 2.2.2 项下方法制备供试品溶液,再按2.2.1 项下色谱条件进样测定,记录峰面积,并计算含量。结果黄芪甲苷含量平均值为0.031 35 mg/mL,RSD为 2.90%(n =6),表明方法重复性良好。
加样回收试验:取已知含量的样品(批号为160501)适量,共6 份,分别加入一定质量浓度的黄芪甲苷对照品溶液,按2.2.2 项下方法制备供试品溶液,再按2.2.1 项下色谱条件进样测定,记录峰面积,并计算回收率。结果见表1。
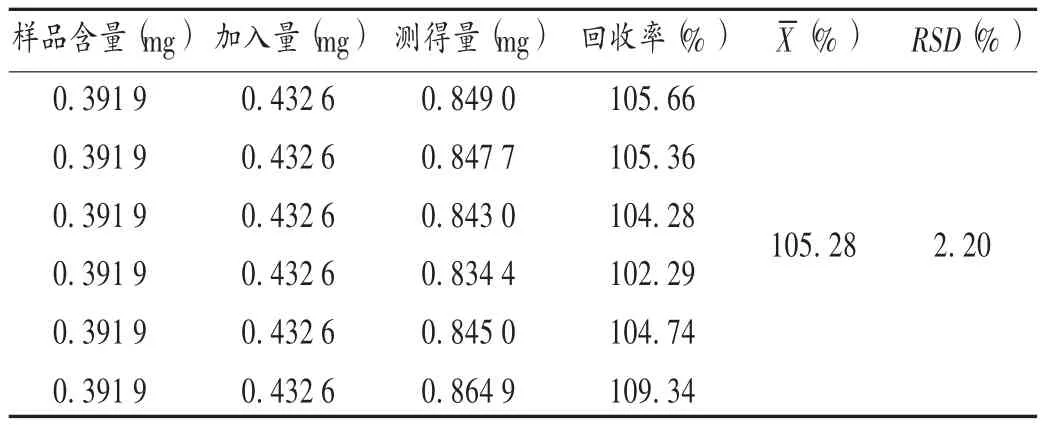
表1 加样回收试验结果(n =6)Tab.1 Results of recovery tests(n = 6)
2.2.4 样品含量测定
取3 批样品各适量,分别按2.2.2 项下方法制备供试品溶液,再按2.2.1 项下色谱条件进样测定,平行测定3 次,记录峰面积,并计算样品含量。结果见表2。
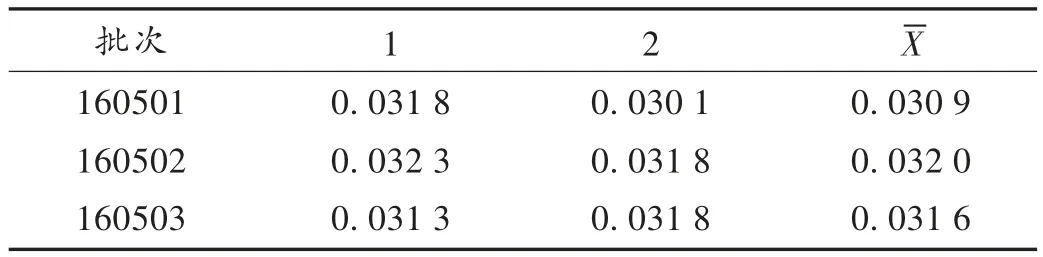
表2 样品中黄芪甲苷含量测定结果(mg/mL,n =2)Tab.2 Results of content determination of astragaloside Ⅳ in the samples(mg /mL,n = 2)
3 讨论
预试验中比较了样品加乙醚振摇及样品浓缩加硅藻土,以甲醇、水饱和的正丁醇分别加热回流提取样品前处理方法,结果均出现斑点分离不佳、拖尾严重或提取过程乳化较严重的现象。参考文献[15]考察展开剂,黄芪TLC 鉴别中考察了三氯甲烷 -甲醇 -水(30 ∶10 ∶1,V / V / V)和三氯甲烷 - 甲醇(10 ∶1,V / V),茯苓 TLC 鉴定考察了石油醚(60 ~90 ℃ )-乙醚(3 ∶2,V / V)和石油醚(60 ~ 90 ℃ )- 乙酸乙酯(1 ∶1,V / V),白术 TLC 鉴别中考察了石油醚(60 ~90 ℃ )-乙酸乙酯(5 ∶1,50 ∶1,V / V),山药 TLC 鉴定考察了正丁醇 -冰醋酸 -水(4 ∶1,V / V)和乙酸乙酯 -甲醇 -浓氨试液(9 ∶1 ∶0.5,V/ V/ V),结果均欠佳。TLC 耐用性分别考察了25 ℃下不同相对湿度(35%,59%,75%),以及相对湿度59%时不同温度(10,25,35 ℃)对 TLC 结果的影响,结果文中所用定性鉴别方法耐用性均较好。
黄芪甲苷作为尿毒营养合剂方中君药黄芪的有效成分[16],因其无紫外吸收的结构特性,提取纯化及检测方法较其他成分更复杂。预试验中,供试品溶液制备方法对比了加硅藻土、乙醚、振摇提取结果。色谱条件考察了不同厂家 C18柱,不同流动相[乙腈 - 水(32 ∶68,V / V)和0.1%甲酸(冰醋酸)-乙腈等]的流动性,不同柱温(30,35,40 ℃),不同漂移管温度(50,60,70 ℃)对含量测定结果的影响。结果黄芪甲苷分离度欠佳,阴性样品中某些成分存在干扰,最终建立了文中所用制备方法及色谱条件。
综上所述,该研究所建立的方法可用于尿毒营养合剂的质量控制。
