喹乙醇印迹传感器的制备及其在喹噁啉类药物残留快检中的应用
2021-03-31田景升李东东赵玲钰高文惠
田景升,李东东,赵玲钰,高文惠
(河北科技大学食品与生物学院,河北 石家庄 050018)
喹噁啉类药物是通过化学合成在喹噁啉环2、3位引入不同官能团的一类广谱抗菌促生长剂,可有效地抑制细菌DNA合成,具有良好的抗菌和抗球虫作用;同时可促进蛋白同化、提高饲料转化率、促进动物生长,被广泛应用于畜禽、水产养殖业[1],常见产品结构式如图1所示。其中喹乙醇(olaquindox,OLA)、卡巴氧(carbadox,CBX)已被证明具有不同程度的致畸性、致突变性和致癌作用[2-3]。我国农业部颁布的第2638号公告及250号公告中明确指出,禁止在食用动物中使用OLA和CBX[4-5]。由中国农业科学院兰州畜牧与兽药研究所研制合成的乙酰甲喹(mequindox,MEQ)、喹烯酮(quinocetone,QCT),在保留传统喹噁啉类药物抗菌活性的同时,大大降低了其毒性作用,可用于食品动物养殖中,但有研究表明MEQ、QCT仍具有一定毒性[6-7]。然而仍有不法商家为扩大药物的作用范围,在其他药物中非法添加MEQ、QCT,造成其混乱使用,并且不能执行休药期,造成食品动物中2 种药物的残留。目前对于喹噁啉类药物的检测方法主要有液相色谱法[8-9]、液相色谱-质谱联用技术[10]、电化学法[11]以及免疫层析法[12]等,但由于动物源性样品基质较为复杂,兽药制剂之间的检测方法差距较大,灵敏度不在同一个数量级,而且药物成分之间存在相互干扰,会对检测造成不利影响[13]。因此,为了有效进行食品安全风险监控,确保消费者饮食健康,有必要建立一种快速、灵敏且抗干扰能力强的检测方法对食品中喹噁啉类药物残留进行测定。
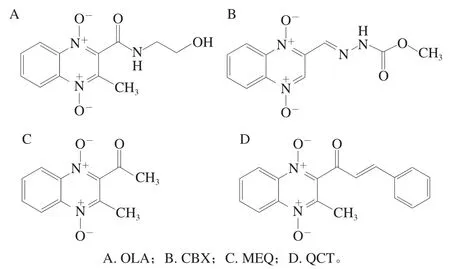
图 1 4 种喹噁啉类药物结构式Fig. 1 Structures of four quinoxalines
分子印迹技术的出现起源于免疫学,其基本原理模仿了生物界的锁-匙原理,类似于抗原-抗体结合[14]。模板分子与功能单体在一定的条件下混合,通过聚合的方式形成具有特异性识别的多重结合位点,再通过浸泡、超声、电位诱导等方式将模板分子去除后,所得分子印迹聚合物(molecularly imprinted polymer,MIP)具有与模板分子大小和形状相匹配的三维空穴,这种印迹空穴对模板分子以及模板分子的结构类似物具有特异选择性[15-16]。并以其独特的结构可预测性、识别特异性和应用广泛性等特点,在环境[17]、食品[18]、药物[19]及天然产物[20]等各领域得到广泛应用。将MIP对目标物质特异性识别的特点与电化学分析快速检测的优势相结合研制出的分子印迹电化学传感器(molecularly imprinted electrochemical sensor,MIECS),由聚合物膜(识别元件)和不同类型的信号转换器组成,利用印迹分子进入印迹膜前后电活性探针或印迹分子本身电化学信号的变化进行检测[21-22]。近年来,随着传感器在电化学领域的不断发展和进步,MIECS已应用于各种食品[23]、环境[24]、药物[25]等领域的分析检测工作中。
虽然分子印迹技术已被广泛应用于目标分子的选择性识别,但传统的MIP制备技术仍存在吸附能力低、扩散阻力大、传质速率慢等不足,因此对分子印迹技术的改进以提高其灵敏度仍是目前的研究热点。研究表明,使用复合功能单体制备的MIP比仅单一功能单体具有更好的选择性[26-27]。复合功能单体含有不同种类的功能基团,模板分子与官能团之间经过聚合之后会形成多种识别位点,可以增强MIP的选择性,同时由于MIP中功能单体之间形成了不同种类的聚合结构交联在一起,可以提高MIP的稳定性。另外采用石墨烯[28]、纳米银[29]、纳米金[30]等材料对工作电极进行修饰,可以增大电极比表面积、增强电极的导电性能。
目前,基于纳米石墨烯与纳米银溶胶作为修饰材料、并采用复合型功能单体制备的OLA分子印迹电化学传感器的制备鲜有报道。本实验以OLA为模板分子,邻氨基苯酚(o-aminophenol,OAP)和间苯二酚(resorcinol,RC)为复合功能单体,通过电化学分析结合紫外光谱法筛选功能单体并优化二者比例,采用滴涂的方式制备纳米石墨烯与纳米银溶胶修饰电极,通过电聚合的方法得到MIP膜,成功制备OLA分子印迹电化学传感器,并应用于食品中喹噁啉类药物快速检测。
1 材料与方法
1.1 材料与试剂
鸡蛋、猪肉、鲽鱼、鲜虾 市购。
OLA(纯度98%)、CBX(纯度98%)、MEQ(纯度98%)、QCT(纯度99%)、邻苯二胺(o-phenylenediamine,OPD)(纯度99%)、对巯基苯胺(4-aminothiophenol,4-ATP)(纯度98%)、RC(纯度99%)、OAP(纯度98%)、石墨烯(纯度>99.5%)、壳聚糖(chitosan,CS)(脱乙酰度≥95%)美国阿拉丁试剂有限公司;硝酸银(纯度≥99.8%)、N,N-二甲基甲酰胺(N,N-dimethylformamide,DMF)、磷酸氢二钠、磷酸二氢钾、铁氰化钾、浓硫酸、柠檬酸三钠、乙酸钠、氢氧化钠(均为分析纯) 成都欣正通化工有限公司;实验用水为超纯水。
1.2 仪器与设备
CHI660E型电化学工作站、三电极系统(CHI104玻碳电极、CHI115铂丝电极、CHI111 Ag/AgCl电极)上海辰华仪器有限公司。
1.3 方法
1.3.1 柠檬酸三钠还原制备纳米银溶胶
根据Lee等[31]的报道,通过改进最常用的柠檬酸三钠还原的方法制备纳米银溶胶。将0.008 g硝酸银固体加水溶解后定容至50 mL,超声混匀;将配制好的硝酸银溶液,加热搅拌至微沸时迅速加入1.5 mL 1%柠檬酸三钠溶液,保持沸腾并持续搅拌,当溶液颜色变为亮黄色时停止加热,冷却至室温,得到纳米银溶胶,密封避光保存。
1.3.2 石墨烯修饰剂的制备
取0.02 g石墨烯加入10 mL DMF,经搅拌、超声分散后与20 g/L的CS-1% CH3COOH溶液等体积混合,磁力搅拌30 min制得石墨烯修饰剂,密封冷藏保存。
1.3.3 紫外光谱法考察功能单体及与模板分子的比例
1.3.3.1 模板分子与不同功能单体的紫外光谱测定
配制5 mmol/L OLA溶液、10 mmol/L功能单体(OAP、RC、OPD、4-ATP)乙腈溶液。取OLA溶液及4 种功能单体乙腈溶液各50 μL分别定容至10 mL;另外固定OLA溶液50 μL,分别配制OLA与4 种不同功能单体浓度比为1∶2的混合溶液,以及将4 种功能单体两两组合分别与OLA溶液配制成浓度比为1∶2∶2的混合溶液,将上述溶液用乙腈定容至10 mL;将上述溶液超声30 min,冷藏环境中静置11 h,进行紫外扫描。
1.3.3.2 不同比例功能单体与模板分子紫外光谱测定
取1.3.3.1节配制的OLA、OAP、RC溶液,固定OLA溶液50 μL,分别配制模板分子与2 种功能单体浓度比为1∶1∶1~1∶6∶6的6 种不同梯度的混合溶液,用乙腈定容至10 mL,超声30 min,冷藏环境中静置11 h,用紫外光谱扫描不同的混合溶液。
1.3.4 OLA分子印迹电化学传感器的制备
1.3.4.1 电极预处理
玻碳电极按参考文献[32]方法进行预处理。
1.3.4.2 OLA分子印迹电化学传感器的制备
用石墨烯修饰剂/纳米银溶胶复合物对预处理电极进行修饰,滴涂到电极表面,用红外灯烘干,得到石墨烯/AgNO3修饰电极;将修饰电极置于聚合液(5 mmol/L OLA、20 mmol/L OAP、20 mmol/L RC)中,在-0.2~1.2 V电位下循环伏安(cyclic voltammetry,CV)扫描25 圈得到电聚合膜,将聚合后的电极浸入10 mL乙醇-0.4 mol/L NaOH溶液(3∶1,V/V)中洗脱20 min去除模板分子,制得OLA分子印迹电化学传感器,即得到带有特异识别空穴的MIP膜。非印迹电极(non-molecularly imprinted electrochemical sensor,NIECS)作为对照,除不添加模板分子OLA外,其他与印迹电极制备方法一致。
1.3.4.3 电化学检测条件
1.3.5 样品处理
取2 g(±0.01 g)样品(鸡蛋、猪肉、鲽鱼、鲜虾)置于50 mL离心管中,分2 次提取,先加入10 mL乙腈溶液,依次进行振荡、超声、离心(4 000 r/min)10 min,静置后从离心管中取出上清液,将剩余残渣再次加入5 mL乙腈二次提取,将2 次上清液合并,用乙腈定容至20 mL,待测[32-33]。
1.4 数据统计
采用Origin 2018软件对实验数据进行数据分析以及图形处理,每个实验重复测量3 次,取平均值。
2 结果与分析
2.1 石墨烯修饰条件的选择与优化

表 1 石墨烯在不同分散剂中分散效果Table 1 Dispersibility of graphene in different dispersants
比较不同分散剂对石墨烯的分散效果以及在电极表面的附着与分布效果,结果如表1所示。单独使用CS-1% CH3COOH溶液作为分散剂时,石墨烯的分散颗粒大小不均一;DMF单独分散石墨烯时,因受其物理特性影响,修饰液在电极表面附着效果不佳。因此,实验采用分散效果稳定的DMF将石墨烯分散后,所得分散液再与附着性良好的CS-1% CH3COOH溶液等体积混合得到石墨烯修饰剂。将适量石墨烯修饰剂滴涂到电极表面,修饰效果较好。
为使石墨烯修饰剂在电极表面稳定修饰且不影响石墨烯的导电作用,实验固定分散剂用量,对石墨烯用量进行优化。通过比较不同石墨烯用量(5、10、20、30、40 mg)所修饰电极的电流响应,以考察石墨烯用量对修饰效果的影响,结果如图2所示。随着石墨烯用量增加,CV曲线氧化还原峰电流强度明显增加,说明电极经石墨烯修饰可提高其电导性;在石墨烯用量为20 mg时,峰电流最高,这时氧化与还原峰电流比接近1∶1;当石墨烯用量超过20 mg,电流强度呈下降趋势,其原因是石墨烯用量超出了所用分散剂的分散能力,致使修饰剂中的石墨烯产生团聚而降低其电导性,另外石墨烯用量过多还会使修饰膜厚度增加影响电流响应。

图 2 石墨烯修饰剂用量的优化Fig. 2 Optimization of graphene dosage
2.2 还原剂和纳米银溶胶的优化

图 3 柠檬酸三钠用量(A)和纳米银溶胶质量浓度(B)的优化Fig. 3 Optimization of trisodium citrate dosage (A) and nanosized silver sol concentration (B)
由于还原剂用量会直接影响纳米银溶胶形成的速度,实验首先优化还原剂柠檬酸三钠的用量。分别考察0.5、1、1.5、2、2.5、3 mL的1%柠檬酸三钠溶液,对修饰效果的影响。如图3A所示,随着柠檬酸三钠用量增加,电极经纳米银溶胶修饰前后其电流响应增幅逐渐增大,当柠檬酸三钠用量为1.5 mL时,响应电流增幅几乎达到最大,继续增加还原剂用量响应电流增幅趋于稳定,说明1.5 mL的柠檬酸三钠可使所配制的硝酸银溶液中的银离子充分还原,且当柠檬酸三钠为1.5 mL时修饰在电极上效果最佳,因此实验确定柠檬酸三钠的用量为1.5 mL。考察质量浓度分别为0.10、0.12、0.14、0.16、0.18、0.20 g/L的纳米银溶胶对修饰效果的影响。如图3B所示,修饰电极的峰电流响应信号随着AgNO3溶液质量浓度逐渐增加而增大;当质量浓度达到0.16 g/L时,峰电流值达到最大。当质量浓度超过0.16 g/L后,纳米银粒子的还原达到饱和,峰电流不再增加。综上,纳米银溶胶的最佳质量浓度为0.16 g/L。
2.3 OLA分子印迹电化学传感器制备条件的优化
2.3.1 功能单体的选择和模板分子与功能单体的比例优化

图 4 OLA与OAP-RC及其混合体系的紫外吸收光谱Fig. 4 Ultraviolet absorption spectra of OLA, OAP-RC and their mixed system
OLA作为喹噁啉氧化物类物质,其分子结构上含有羰基、胺基、羟基等极性基团较多,因此考虑采用氢键作用力这种非共价键的形式组装模板分子与功能单体。使用紫外光谱法结合电化学分析,在4 种含有极性基团的常用功能单体及其两两组合的6 种复合型功能单体中进行筛选。通过测定OLA与不同功能单体之间紫外吸光度理论值与实测值之差ΔA,考察各功能单体与OLA之间氢键作用力的大小。单一功能单体与OLA的ΔA分别为OAP:0.352 2、RC:0.349 3、OPD:0.232 8、4-ATP:0.347 1。复合功能单体与OLA的ΔA分别为OAP-RC:1.182 4、OAP-OPD:1.177 6、OAP-4-ATP:1.180 5、RC-OPD:1.176 6、RC-4-ATP:1.142 3、OPD-4-ATP:1.031 5。比较上述结果可知OLA与OAP-RC之间的ΔA最大,结果如图4所示,因此可判断OLA与OAP-RC之间的作用力最大。因此实验选择OAP-RC为最佳复合功能单体。

图 5 模板分子与功能单体比例的优化Fig. 5 Optimization of ratio of template molecules to functional monomers
聚合体系中模板分子与功能单体之间的比例是影响MIP膜上印迹识别位点的关键因素之一,若功能单体浓度过低会减少印迹位点的形成,降低MIP膜的选择性和传感器的抗干扰能力;浓度过高则会使功能单体无规则缔合,降低检测灵敏度[34]。因此需要对功能单体与模板分子比例进行优化。实验采用紫外光谱法进行扫描,当最大吸收波长对应的吸光度趋于平缓时,说明模板分子与功能单体之间的作用趋于稳定,此时平缓区的转折点作为最佳比例。由图5可知,当OLA与OAP、RC的比例在1∶4∶4之前吸光度的变换稳定上升,在浓度比超过1∶4∶4后吸光度变化趋于平缓。由此推断二者的最佳比为1∶4∶4。
2.3.2 聚合电解质及其pH值的选择

图 6 不同电解质聚合峰电流曲线Fig. 6 Polymerization peak current curves of different electrolytes
实验选取PBS、醋酸钠、氯化钾、氯化铵4 种常用的聚合电解质溶液,在相同聚合条件下,使复合功能单体OAP-RC和模板分子OLA分别在4 种电解质溶液(pH 7.0)中进行电聚合,通过CV测量在不同电解质溶液中聚合后印迹电极的电流响应,以研究不同聚合电解质溶液对聚合过程产生的影响。如图6曲线a所示,PBS作为电解质溶液时,响应峰电流最高,峰电位较小,且洗脱模板分子后效果最佳,可形成较多稳定的印迹位点,因此实验选择PBS作为最佳电解质溶液。

图 7 电解质pH值的优化Fig. 7 Optimization of electrolyte pH
电解质溶液pH值会影响模板分子与功能单体之间的相互作用,进而对聚合反应产生影响。实验考察pH 5.0、6.0、7.0、8.0、9.0、10.0、11.0、12.0的PBS溶液对聚合反应影响,结果由图7所示,当pH 7.0时,聚合后响应峰电流最大,成膜最稳定,聚合效果最好。综上所述,最佳聚合电解质溶液为PBS(pH 7.0)。
2.3.3 聚合电位和聚合圈数的选择
根据模板分子和功能单体相互作用的成膜电位,考察-0.2~1.4、-0.2~1.2、-0.2~1.0、0~1.2、0~1.4 V 5 种不同电位范围对聚合效果的影响。实验结果显示,在0~1.4、0~1.2、-0.2~1.0 V电位范围进行聚合后,所形成的聚合物膜不稳定,在洗脱过程中出现了不同程度的脱落现象;当电位在-0.2~1.2 V范围时聚合物膜稳定,洗脱效果良好;进一步扩大电位范围至-0.2~1.4 V时,出现模板分子洗脱困难的现象,其原因是电位过高会加强模板分子与功能单体之间作用力的强度,使得模板分子难以洗脱;所以选择-0.2~1.2 V的电位范围作为最佳聚合电位。

图 8 聚合圈数的优化Fig. 8 Optimization of number of polymerization circles
聚合物膜的薄厚,会对检测结果产生直接影响[35]。聚合物膜过薄,所形成印迹位点较少、选择性低、抗干扰能力弱且膜不稳定易脱落;聚合物膜过厚会使模板分子被包埋在MIP膜内,造成洗脱困难等影响,降低传感器灵敏度。因此聚合过程中常通过调整扫描圈数实现控制聚合物膜厚度的目的,结果如图8所示,随着聚合圈数的增加,响应电流不断增大,在聚合20 圈时响应峰电流最高,继续增加聚合圈数,响应电流降低。亦即聚合20 圈时,聚合物膜所形成的印迹位点较多;超过20 圈后模板分子因聚合物膜过厚而出现被包埋而难以洗脱的现象,导致无法形成更多的印迹位点,降低了灵敏度。综上,确定聚合圈数为20 圈。
2.4 分子印迹膜的聚合过程
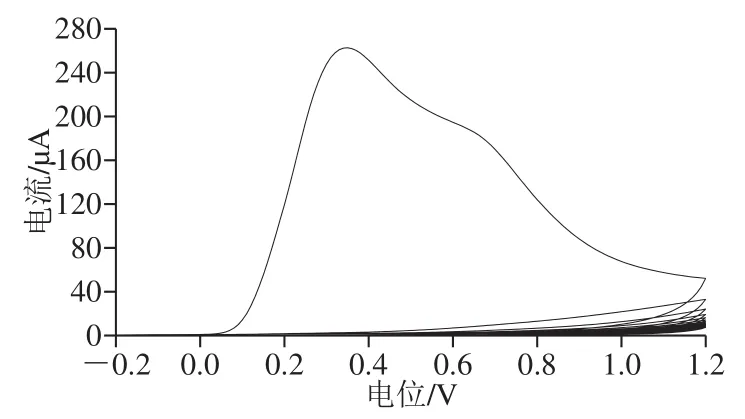
图 9 MIP膜的电聚合曲线Fig. 9 Electro-polymerization curve of MIP membrane
实验采用CV法进行电聚合,由图9可以看出,MIP形成过程不可逆。聚合第1圈时氧化峰出现在0.347 V,且峰电流达到263 μA;当聚合第2圈后,峰电流值迅速降低,随着聚合圈数的增加,峰电流逐渐降低并趋近于0,表明此时形成了致密稳定且不导电的聚合物膜。将聚合后的电极放置于探针离子溶液中表征,由于电子转移受到聚合膜的阻碍,几乎没有氧化还原峰电流出现,再将分子印迹膜置于洗脱剂中洗脱去除模板分子后,得到带有印迹空穴的MIP膜。
2.5 模板分子洗脱条件的选择与优化

图 10 NaOH浓度(A)和洗脱时间(B)对洗脱效果的影响Fig. 10 Optimization of NaOH concentration (A) and elution time (B)
选取0.5 mol/L H2SO4-50%甲醇(1∶1,V/V)、0.5 mol/L HCl-50%甲醇(1∶1,V/V)、0.5 mol/L H2SO4-50%乙醇(1∶1,V/V)、0.5 mol/L HCl-50%乙醇(1∶1,V/V)、乙醇-0.4 mol/L NaOH溶液(3∶1,V/V)作为洗脱剂,将聚合好的电极分别置于上述洗脱剂中浸泡洗脱20 min后表征发现,在酸性溶液中难以破坏模板分子与功能单体间的氢键,致使OLA不易被洗脱,形成的印迹空穴较少,峰电流响应信号弱;当电极置于碱性溶液中洗脱后表征出现了氧化还原峰电流,表明此时模板分子已从聚合物膜中洗脱形成印迹空穴。为了获得更好的洗脱效果,优化了洗脱液中的NaOH浓度。由图10A所示,随着NaOH浓度的增加,洗脱后氧化峰电流逐渐增大,当浓度超过0.4 mol/L时,洗脱模板分子后峰电流异常增大,这是由于NaOH浓度过高破坏了分子印迹膜而使其脱落。因此,选择乙醇-0.4 mol/L NaOH溶液(3∶1,V/V)作为最佳洗脱剂。由图10B可以看出,随着洗脱时间的不断延长,洗脱后的电极响应峰电流逐渐增大,当洗脱时间达到20 min后,峰电流值趋于稳定,因此模板分子的最佳洗脱时间为20 min。
2.6 传感器吸附性能考察
2.6.1 静态吸附曲线
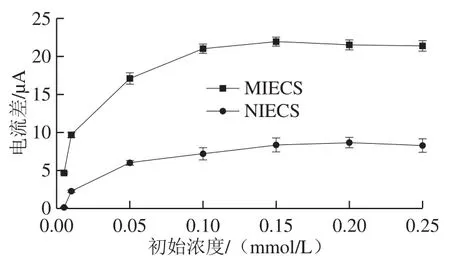
图 11 静态吸附曲线Fig. 11 Static adsorption curves
将MIECS与NIECS分别在不同浓度的OLA标准溶液中静态吸附10 min后,表征吸附后响应电流值,如图11所示。吸附后MIECS的电流变化远高于NIECS,其原因是MIECS特异吸附OLA后,其上的印迹空穴被OLA占据,阻碍了探针离子穿过聚合物膜发生氧化还原反应,使吸附前后电流响应产生较大的变化;而NIECS吸附前后的电流变化很小,可见NIECS对OLA的吸附量很小,说明NIECS未形成大小形状与OLA互补的印迹空穴,不能产生特异性吸附作用,而仅以物理吸附的方式吸附少量OLA。
2.6.2 吸附动力学曲线

图 12 吸附动力学曲线Fig. 12 Adsorption kinetic curves
将MIECS与NIECS分别在0.1 mmol/L的OLA标准溶液中进行动态吸附,每2 min测定一次电流响应,结果如图12所示。MIECS随着吸附时间的延长,氧化峰电流逐渐减小,吸附10 min后峰电流不再下降,表明MIECS上的印迹空穴几乎全部被OLA占据,阻碍了探针离子的氧化还原,此时MIECS吸附至饱和;相比之下NIECS的电流变化较为缓慢,直至吸附14 min后变化趋于平缓,说明NIECS对OLA的吸附能力远小于MIECS,进一步验证了NIECS未形成形状大小与OLA互补的印迹空穴。
2.7 传感器的印迹效应
实验通过SWV表征MIECS的印迹效应。由图13可以看出,石墨烯与纳米银溶胶修饰电极后氧化峰电流达到最大,曲线a表明电极修饰效果良好;聚合后的电极几乎没有出现氧化峰,曲线d表明此时功能单体之间以及模板分子与功能单体相互作用,在修饰电极表面形成了一层近乎绝缘的聚合物膜;当电极置于洗脱剂中浸泡洗脱模板分子后,再一次出现氧化峰响应信号,曲线b说明此时模板分子已被去除得到印迹空穴,进而可使探针离子在电极表面发生电子转移;将洗脱模板分子后的电极重新置于模板分子OLA标准溶液中吸附后发现,氧化峰电流值再次降低,曲线c表明部分模板分子又重新占据到印迹空穴表面,阻碍了电子转移。以上结果分析,该传感器具有良好的印迹效应。
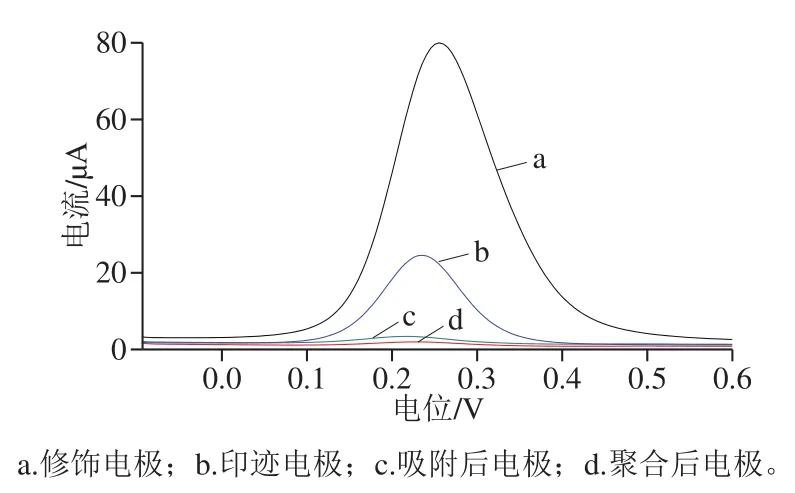
图 13 不同电极在探针溶液中的SWV曲线Fig. 13 SWV curves of different electrodes in probe solution
2.8 传感器的选择性

图 14 印迹传感器的选择性Fig. 14 Selectivity of the sensor
实验对模板分子OLA在内的4 种喹噁啉类药物以及与喹噁啉类结构相差较大的氯霉素作为干扰物质对印迹传感器的选择性进行考察。将制备好的印迹电极分别置于0.1 mmol/L的5 种目标物质的标准溶液中浸泡进行吸附性实验,并采用SWV法对吸附前后的氧化峰电流变化值进行测定。通过图14中氧化峰电流变化值可以看出,传感器对模板分子OLA的氧化峰电流变化值最大,对其结构类似物CBX、MEQ、QCT的氧化峰电流变化值有所降低,对干扰物质几乎没有峰电流响应。其原因是CBX、MEQ、QCT与MIP膜上印迹空穴的大小、形状、识别位点不能完全契合,相比于OLA进入印迹空穴的量有所减少,因此吸附前后的氧化峰电流变化值小于OLA;而氯霉素的结构与OLA相差较大,难以进入印迹空穴,故其氧化峰电流变化值小。该结果表明传感器对4 种喹噁啉类药物均具有特异识别作用,其中对OLA的选择性最高,表明该传感器抗干扰能力强。
2.9 印迹传感器的分析性能与应用
2.9.1 线性关系与检出限

图 15 4 种不同浓度的目标物质线性关系Fig. 15 Linear relationship curves of four target analytes at different concentrations
实验在最优条件下,分别配制了OLA、CBX、MEQ、QCT的不同浓度梯度标准溶液(0.003 μmol/L~0.8 mmol/L),通过SWV法4 种目标物质在其不同浓度标准溶液中吸附前后的峰电流变化值进行测定。以氧化峰电流变化值(ΔI)为纵坐标,标准溶液浓度的对数值(-lgC)为横坐标作图考察该传感器对4 种目标物的线性关系。由图15可以看出,模板分子及其结构类似物均在一定浓度范围内呈良好的线性关系,如表2所示,模板分子OLA的线性方程为ΔI=-1.344 9lgC+12.766,R2=0.999 1,检出限为1.0×10-9mol/L。结果表明该MIECS能够满足4 种喹噁啉类药物的检测需求。
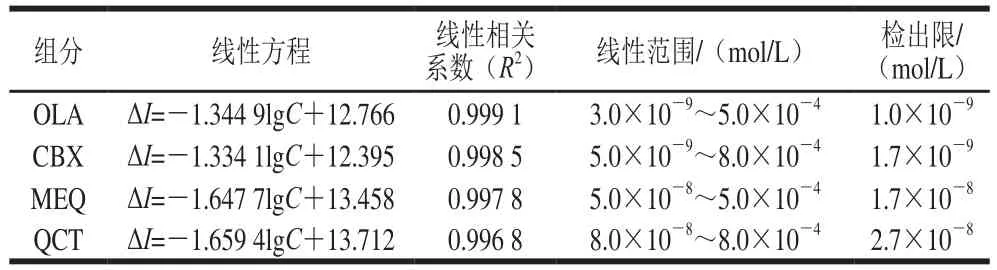
表 2 4 种目标物的线性关系及检出限Table 2 Linear calibration and detection limits of four target analytes
2.9.2 回收率与精密度实验结果
在最优条件下,分别在3 个添加水平下进行样品加标回收率实验,结果如表3所示。OLA样品加标平均回收率在89.05%~100.86%之间,相对标准偏差(relative standard deviation,RSD)在1.03%~3.11%之间(n=5),表明该快速检测方法回收率与精密度良好。

表 3 样品加标回收率和精密度(n=5)Table 3 Recoveries and precision for spiked samples (n= 5)
2.9.3 重复性与稳定性结果
实验平行制备3 支印迹传感器通过SWV连续洗脱吸附20 次,考察传感器的重复性。对模板分子OLA标准溶液洗脱吸附后,该3 支传感器的峰电流响应信号RSD分别为1.57%、1.64%、1.91%,表明电极重复性良好;将制备好的3 支印迹电极置于4 ℃保存,分别放置1、2、3 周后再对电极进行测定,峰电流响应信号RSD分别为1.37%、2.58%、3.09%,表明电极具有良好的稳定性。
2.9.4 实际样品检测及该快检方法与其他文献比较
针对小样本量样品(鸡蛋、猪肉、鲽鱼、鲜虾)进行检测,检测结果为实际样品中未检出上述4 种喹噁啉类药物。
以喹噁啉类药物中OLA的检测为例,将OLA印迹传感器检测方法与国家标准及文献报道的OLA检测方法进行比较,由表4可知,该方法检出限低,线性范围宽。而且该方法抗干扰能力强,检测快速,满足实际样品的检测要求。

表 4 该方法与其他测定OLA的方法比较Table 4 Comparison of this method with other methods for the determination of OLA
3 结 论
本实验基于柠檬酸三钠还原硝酸银制备纳米银溶胶结合纳米石墨烯修饰工作电极,有效提高了其电导性;以OLA为模板分子,通过紫外光谱法结合电化学分析筛选OAP-RC作为复合型功能单体,并对其在聚合体系中所占配比进行考察;采用电聚合法制备MIP膜,优化制备条件:以PBS(pH 7.0)作为支持电解质溶液,在-0.2~1.2 V电位下聚合20 圈;在乙醇-0.4 mol/L NaOH溶液(3∶1,V/V)中洗脱20 min去除模板分子后,成功制备OLA分子印迹电化学传感器;对该传感器的印迹效果和选择性进行考察,该传感器印迹效果好,且可特异性识别喹噁啉类药物,有较强的抗干扰能力;该方法对喹噁啉类药物线性范围宽,检出限在1.0×10-9~2.7×10-8mol/L之间,OLA样品加标回收率为89.05%~100.86%,RSD不高于3.11%。该传感器具有良好的重复性和稳定性,选择性好,可快速测定实际样品中喹噁啉类药物残留。
