铜基片静态气液界面腐蚀规律的实验
2021-03-30陈宏霞刘霖李林涵
陈宏霞,刘霖,李林涵
(华北电力大学能源动力与机械工程学院,北京102206)
气液两相流动在能源、化工、石化、核工业以及环境工程等工业中广泛存在,一旦两相流存在腐蚀粒子或相界面剧烈冲击都会导致严重后果。如气液热交换器[1-2]、涡轮叶片[3]、气井采集[4]和运输管线[5]常因气液相腐蚀引起停工停产甚至重大事故。但对于气液相界面的腐蚀机理仍有待深入研究,以便实现对界面腐蚀更准确地预测、监测及控制。
基于经典的腐蚀基础研究,近年来对气液界面腐蚀的研究主要可分为空蚀、水线腐蚀、露点腐蚀等几类。Soyama[6]利用孔板向水中注入高速水射流构建水动力空化气泡,研究气泡在液相中的腐蚀行为,结果证明在一定时间内气泡溃灭在金属表面会产生氧化层,并且该氧化层与直接浸没在溶液中产生的氧化层不同,是一种新的N—O化合物。除此之外,Luo等[7]、Yong等[8]也开展了空蚀研究,研究气泡动力学的影响获得了类似的结论。陈亚林等[9]、邢佩等[10]、Li 等[11]利用阵列电极精确定位并监测金属电极气液界面处的水线腐蚀,获得各微电极表面的腐蚀电位和腐蚀电流变化规律,证明线腐蚀界面的波动及界面腐蚀与浓度相关的扩散腐蚀机理,阵列电极的引入可精准测定非固定相界面,证明了线界面的波动特征。Ebara 等[12]和Ding 等[13]针对余热锅炉的炉管、省煤气管中的露点腐蚀,研究金属壁面经一段时间冷凝换热后的腐蚀产物形貌和结构推测露点腐蚀的基本反应方程式。
空蚀[14]主要特点表现为气泡随机产生,且其界面腐蚀耦合了气泡冲击的过程;露点腐蚀[15-16]以及水线腐蚀[17-21]同样主要以金属壁面上的气液界面为研究对象,相比空蚀其相界面可在金属壁面驻停较长时间。不同种类的界面腐蚀存在差异的同时具有显著的共同点,即腐蚀相界面不稳定且不断演变,这一特征使得研究界面腐蚀机理难度加大;因此,对于空泡和水线的界面腐蚀的研究多通过长时间的浸泡获得多点叠加的腐蚀规律[6-8],对于露点腐蚀则主要获得伴随液体体积及液滴内浓度变化的腐蚀速度规律[22-26],再基于金属表面腐蚀产物形貌、成分的检测解释其腐蚀机理[10]。显而易见,要保证界面稳定、气液浓度稳定,将纯粹界面对腐蚀的影响从总面积、长时间叠加的腐蚀规律中独立剥离出来较难,但只有真正剥离界面的作用规律才能从机理上认识相界面腐蚀,其关键在于控制稳定的界面。
本文利用向倒置在溶液中的铜片表面注入一定体积的气泡,维持气泡在液体内的稳定存在保证了界面的稳定以及气液两相内离子浓度的恒定;采用电化学方法对比研究有无稳定气液界面的静态腐蚀规律,真正剥离了相界面的影响,为对界面腐蚀准确建模、预测腐蚀速率以及进一步深入研究奠定基础。
1 实验系统
静态腐蚀实验台主要包括浸泡腐蚀系统和检测系统。浸泡腐蚀系统由高透明度可视玻璃加工而成的储液槽(92mm×60mm×80mm)、用于控制气泡体积的PLC步进电机系统、导气管及出气针头(直径为0.64mm)组成;检测系统则是由经典的三电极体系及相应数据采集电化学工作站系统组成,如图1所示。本文中工作电极为20mm×20mm×2mm的铜片,测试面积为20mm×20mm;依次采用1500#、2000#的砂纸逐级打磨、乙醇清洗后待用。4cm×4cm 的铂片作对电极,Ag-AgCl 标准电极作参比电极。
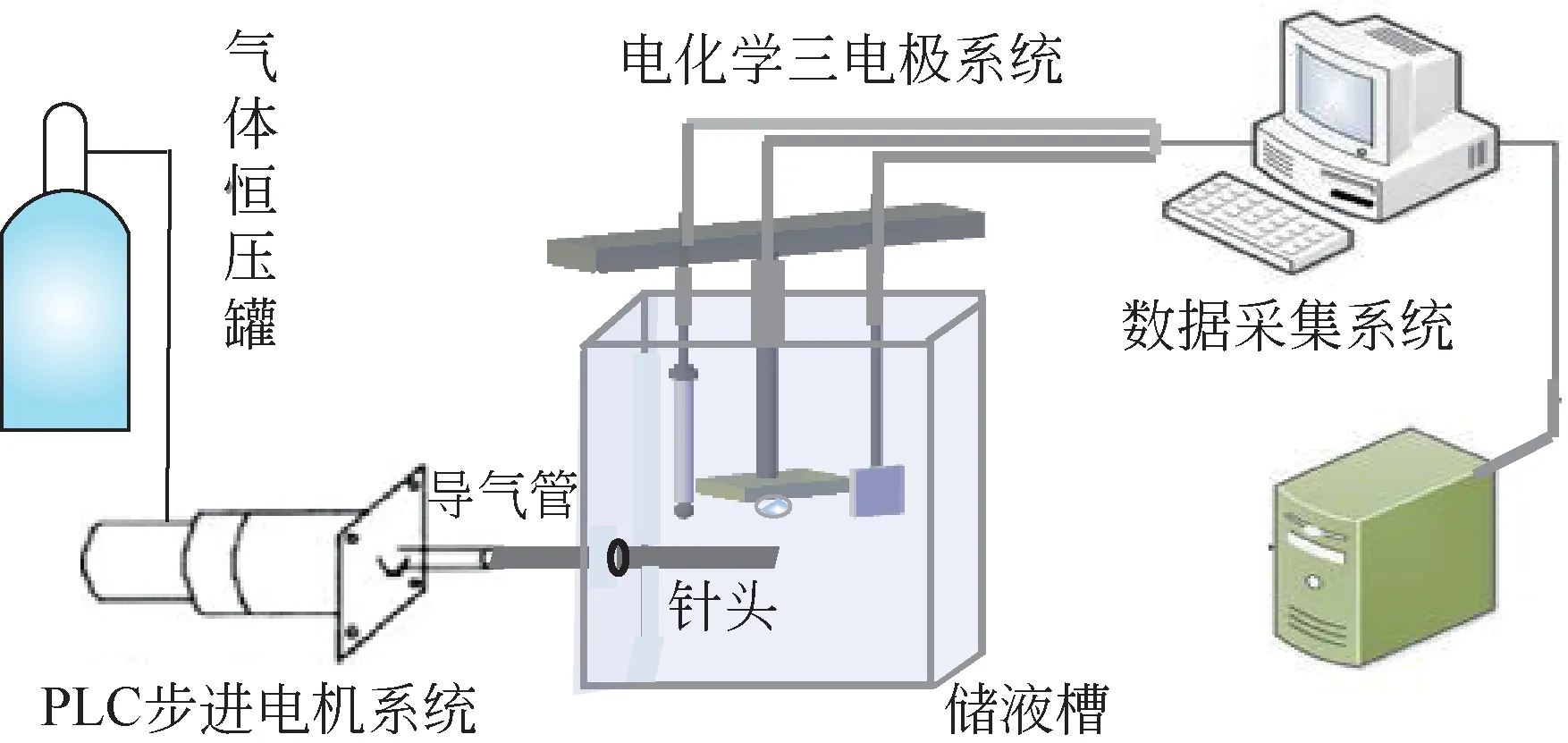
图1 实验装置示意图
本 文 针 对0.1mol/L、 0.2mol/L、 0.3mol/L、0.6mol/L、0.8mol/L、0.9mol/L不同浓度NaCl溶液下有无气液界面处腐蚀行为进行对比研究。进行电化学测试时,动电位极化曲线扫描的范围为-500~0mV,扫描速率为1mV/s。电化学阻抗谱的测量是在试样于测试溶液中的开路电位下进行的,施加的正弦波幅值为10mV,扫描频率为0.1~106Hz。
2 实验结果与讨论
2.1 静态界面的腐蚀规律
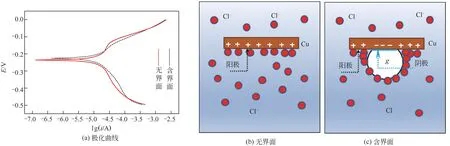
图2 有无界面情况下金属铜的腐蚀规律
图2 为有无界面条件下铜在NaCl 溶液(浓度c=0.2mol/L,pH=7,温度t=20℃)中的腐蚀规律,可知极化曲线在有无界面时整体形状相似,两种情况下金属试样均未出现明显钝化。根据自腐蚀电位比较可知,引入稳定气液界面后,金属的自腐蚀电位从-237mV 略微升高至-223mV,说明引入气液界面后在某种程度上抑制了金属表面腐蚀反应的发生。如图2(b)所示,无气液界面时整个金属表面浸泡在除氧后的溶液中,其金属表面主要发生金属溶解的阳极反应。而当引入气泡使金属表面维持稳定气液界面时,金属表面被划分为两个区域,即气泡覆盖表面和固-液接触表面,如图2(c)所示。气泡由含氧量21%的空气组成,气泡覆盖金属表面的微液层厚度较薄,促进了阴极反应过程中氧的扩散和对流;同时较薄的微液层使阳极反应受液相传质过程控制导致气泡内Cu 金属的溶解反应受到抑制[27]。而气泡外金属表面为少氧的离子腐蚀,两区域表面不同的反应及反应电势使得在金属表面形成新的微电池;根据两侧的反应电势可知气相区为微电池的阴极,溶液浸没区为微电池的阳极[18,28]。气泡内阴极表面金属发生的溶解反应在微电池出现后被抑制,使引入气液界面的Cu 表面自腐蚀电位升高。
需指出的是,自腐蚀电位升高只说明腐蚀发生的难易程度,一旦腐蚀发生后腐蚀速率的大小则由腐蚀电流来表征。对图2极化曲线中的强极化区进行拟合计算,可知含界面试样的腐蚀电流密度(1.4528×10-6A/cm2)高于无界面试样的腐蚀电流密度(1.3808×10-6A/cm2),即引入气液界面虽然使腐蚀电位升高、腐蚀反应较难发生,但腐蚀一旦发生后金属表面腐蚀电流密度增大。因为除了气泡外与溶液接触的金属发生溶解反应,气泡内外具有较大的电势差,此界面处电势差下的微电池反应及金属表面的腐蚀速率具有叠加效应,使腐蚀电流密度增大、腐蚀更剧烈。
另一方面,由于气液界面张力及Maragoni效应的作用,引入气液界面后使得液相离子浓度有所降低,液相主体区部分Cl-汇集在气液界面附近的一薄液膜层[29];气泡内氧同样在界面发生向液相主体区的浓度扩散,且同样在界面附近的薄层内具有较高浓度。因此,引入界面后腐蚀最严重的区域往往发生在离子浓度最高、氧浓度最高的界面区。
2.2 Cl-浓度对静态界面腐蚀规律的影响
2.2.1 极化曲线
气液界面的腐蚀离不开界面薄层内Cl-的扩散和氧分子的扩散,本节通过对比多组不同浓度下有无界面的腐蚀参数进一步探究界面腐蚀规律。
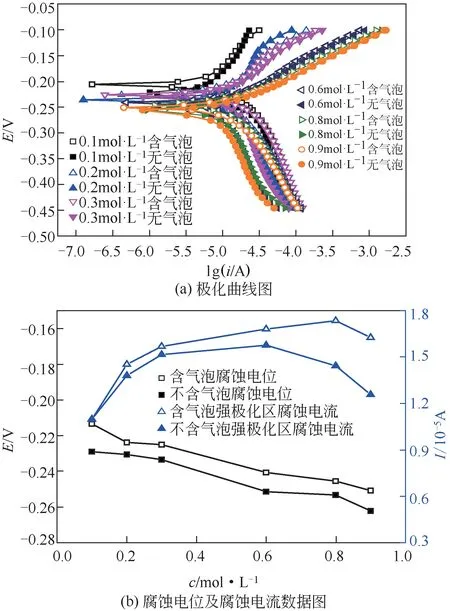
图3 腐蚀电位、腐蚀电流随Cl-浓度的变化曲线图
图3(a)为不同溶液浓度下有无稳定气液界面时Cu表面的极化曲线图,并对曲线中的腐蚀电位以及强极化区腐蚀电流密度进行分析计算,获得图3(b)所示的腐蚀电位、强极化区腐蚀电流密度随Cl-浓度变化曲线图。可知,在Cl-浓度从0.1mol/L 升高至0.9mol/L范围内,引入稳态气液界面后铜试样的腐蚀电位均发生正移且升高幅度较为一致,表明该浓度范围内引入气液界面后均在金属腐蚀表面引入微电池,降低其受腐蚀的倾向;同时,微电池内电流传递均使具有稳定界面的铜试样表面腐蚀电流密度升高,且强极化区电流密度增大显著,浓度为0.9mol/L溶液下强极化区电流密度增大幅值可达。此外,强极化区腐蚀电流密度随溶液浓度的升高呈现先增大再减小的变化规律,即溶液中Cl-浓度的升高首先增大了腐蚀速率,而当浓度升高到一定程度后腐蚀离子浓度反而抑制了腐蚀反应的进行或在高浓度下腐蚀离子浓度不再为控制腐蚀反应的关键因素。
2.2.2 界面腐蚀速率
无界面条件下的铜表面腐蚀实验后产生较均匀的离子腐蚀,整体颜色加深。含界面条件下的铜表面在气泡附着处留下了较为清晰的腐蚀环线圈,如图4所示。圈外液相区域的铜表面颜色加深、腐蚀严重;而圈内区域的颜色与实验前用砂纸打磨过的铜表面相比几乎无变化;可知圈内区域含氧腐蚀为微电池的阴极,其金属溶解过程受到抑制,腐蚀速率极低。假设界面内腐蚀速率可忽略,在不考虑气泡内部金属溶解腐蚀速率的情况下,含界面的Cu金属腐蚀速率则由液相腐蚀速率和界面腐蚀速率两部分组成,因此在保持液相腐蚀离子浓度相同的情况下,综合考虑面积占比的影响后对比有无气液界面的腐蚀电流,计算其差值即可定量表达界面腐蚀电流,如式(1)所示。
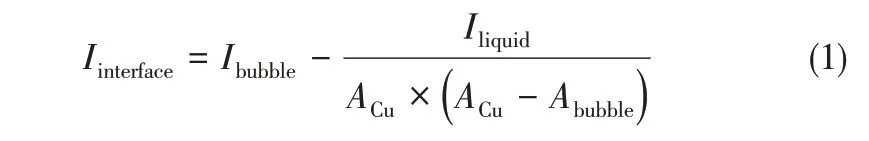
式中,Iinterface为界面腐蚀电流,A;Ibubble为含界面时的腐蚀电流,A;Iliquid为无界面时的腐蚀电流,A;ACu为铜片表面积,cm2;Abubble为铜表面的气泡附着区域面积,cm2。由于在界面薄膜处腐蚀粒子及氧分子浓度高、腐蚀较严重,极化曲线测量过后铜表面上呈现清晰的气泡轮廓线,经过图像处理即可提取腐蚀轮廓线并获得Abubble。通过不同反色处理可得到两个明显的腐蚀环线R1与R2,如图4 所示,R1为相界面液相侧边界,R2为相界面气相侧边界,显然Abubble表示利用R2计算的气泡附着在铜表面的圆形面积,R1与R2之间区域为相界面腐蚀影响的区域。利用实验获得的R2数据即可根据式(1)计算界面腐蚀电流Iinterface。同时利用腐蚀电流、R1与R2确定的界面腐蚀影响面积以及年腐蚀深度的关系可获得界面年腐蚀深度,如式(2)所示。

式中,m 为原子量;n 为得失电子数;D 为金属材料密度,g/cm3。
表1为不同溶液浓度下引入界面后气泡覆盖金属表面的面积及所占份额,表2为不同浓度下界面腐蚀电流及界面年腐蚀深度数据表。可知在整个浓度范围内界面腐蚀速率随着Cl-浓度的升高而增大,表明界面对腐蚀的影响随浓度的升高而愈发明显。
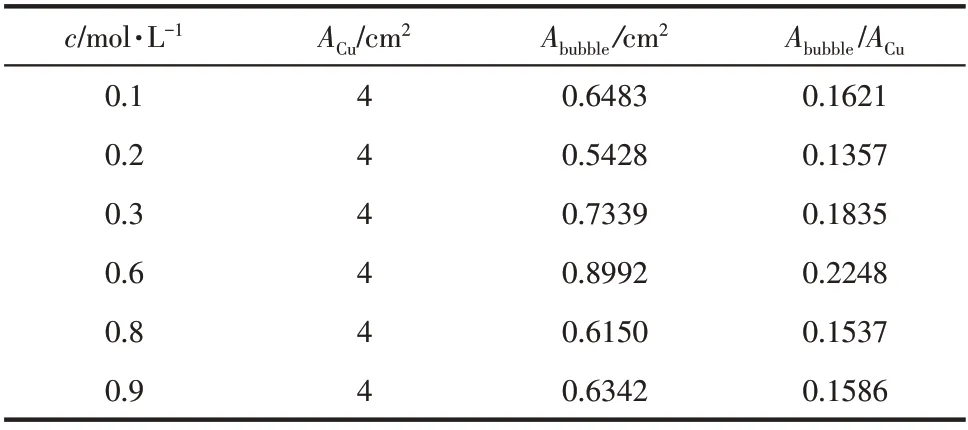
表1 不同浓度下引入界面后气泡覆盖金属表面面积所占份额
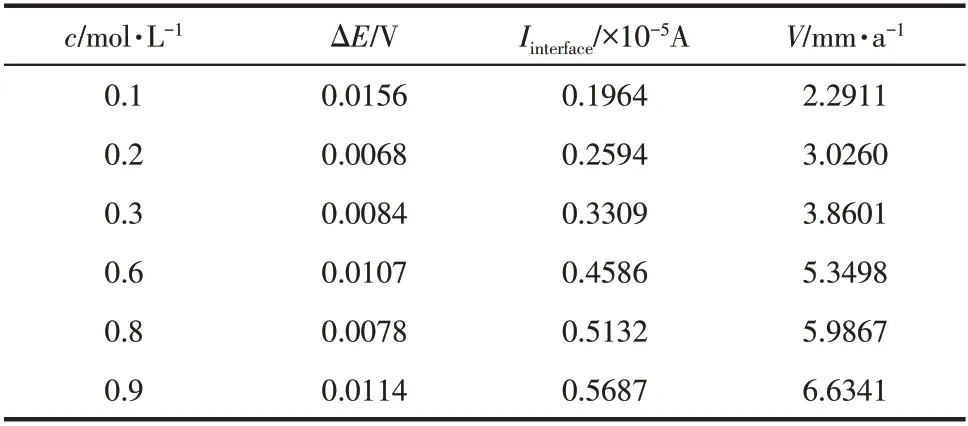
表2 不同浓度下引入界面后腐蚀电势的增幅及界面腐蚀速率

图4 气液界面处腐蚀形貌及腐蚀轮廓线处理
2.2.3 电化学阻抗谱
为深入探究界面腐蚀机理,通过电化学阻抗谱测量对电极动力学过程进行分析。图5(a)所示为在不同浓度的NaCl 溶液(pH=7,t=20℃)中有无界面条件下铜试样的电化学阻抗谱。由图可知,在低浓度(0.1~0.3mol/L)时,阻抗谱由高频区的容抗弧和低频区的直线组成,表明在此范围内的腐蚀过程受电化学反应及浓度扩散混合控制;在高浓度(0.6~0.8mol/L)时,容抗弧彻底消失,腐蚀过程受浓度扩散控制。
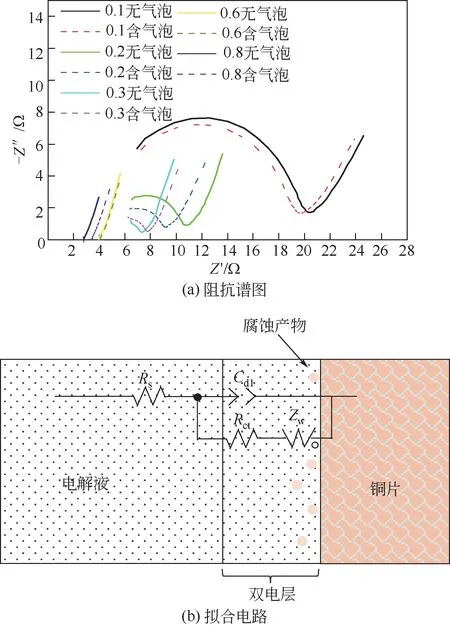
图5 电化学阻抗谱测量结果
采用如图5(b)所示的等效电路模型R[C(RW)]进行分析,其中Rs为参比电极与被测电极之间的溶液电阻(Ω/cm2),Rct为双电层电荷转移电阻(Ω/cm2),Cdl为电极试样与溶液之间的双电层电容(F/cm2),Zw为扩散引起的扩散电阻(Ω/cm2)。利用Zview 软件对低浓度下阻抗谱图进行数值拟合可获得如表3所示的数据结果。首先对比有无界面工况,可知引入气液界面后减小了双电层电荷转移电阻(含 界 面Rct=13.67~2.831Ω/cm2;无 界 面Rct=14.57~2.657Ω/cm2)及扩散电阻(含界面Zw=5.747~2.503Ω/cm2;无界面Zw=4.401~2.86Ω/cm2),因此含界面时的腐蚀总电阻减小,使得腐蚀电流增大。同时,对比不同浓度下的腐蚀速率,可知在低浓度(0.1~0.3mol/L)时,阻抗谱包含明显的容抗弧及扩散直线,表明腐蚀由电化学过程及扩散过程混合控制;在高浓度(0.6~0.8mol/L)时,阻抗谱中容抗弧彻底消失只有扩散直线,表明此时腐蚀全程受扩散过程的影响。计算阻抗谱图中扩散直线的斜率,可知溶液浓度从0.6mol/L 升高至0.8mol/L 时,含界面的试样阻抗谱图中扩散直线斜率从2.264 降低为2.024,无界面试样则从2.399 降到2.167。由上文可知,界面腐蚀主要受界面两侧薄膜内离子浓度控制,同时界面腐蚀的引入能够显著地减小扩散电阻Zw,使得引入界面腐蚀后直线斜率随浓度升高降低的幅度增大,表明在高浓度扩散过程为主要控制机理时界面腐蚀对总腐蚀速率的贡献更为显著。此外,Cl-浓度变化、有无界面对参比电极与被测电极之间溶液电阻Rs影响不大;界面双电层电容Cdl随金属表面腐蚀产物增多电极表面粗糙度增大显示常规增大规律。
2.3 静态气液界面的腐蚀机理分析
界面腐蚀机理离不开界面双膜理论及其相应的界面效应,如图6(a)所示为气液相界面双膜理论示意图,双膜即在相界面两侧存在稳定的气体滞流层(气膜)和液体滞留层(液膜)[30]。经典双膜理论认为,在气液双膜内为气液两相的过渡状态[31]。随时间的变化两侧不同物质粒子不断在浓差作用下发生传递,使膜内气液两相浓度不断发生变化;在此粒子扩散运动过程中宏观界面形状也随之发生细微波动,这在电极阵列研究开放式界面腐蚀过程中已被证实[18]。同时,毛志红[31]通过分子动力学模拟也证实在氧气-水气液界面处两侧存在势垒和势阱,无论氧分子由气相传递到液相扩散或从液相分离进入气相均伴随能量的耗损,如图6(b)所示。因此在浓度扩散及Maragoni作用下界面两侧的双膜内同时存在两相的溶质分子;且相对侧的一相中溶质由于消耗能量进入界面后仍需继续克服能垒方可进入本侧主体,使得对侧溶质离子在膜层界面处均为最高浓度。此界面处离子浓度与主体离子浓度分布具有显著区别,使得金属在气液界面与浸泡主体区域间形成宏观氧浓差电池腐蚀。
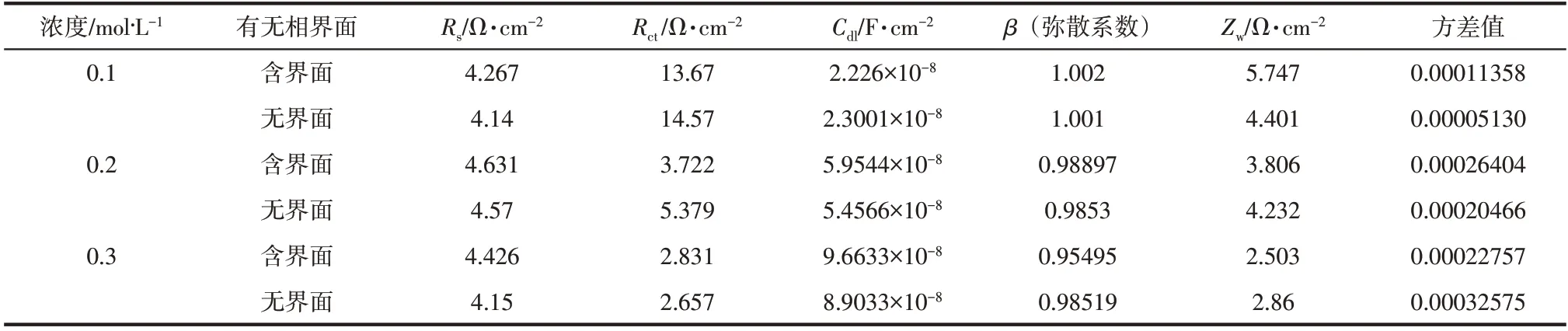
表3 电化学阻抗谱拟合结果
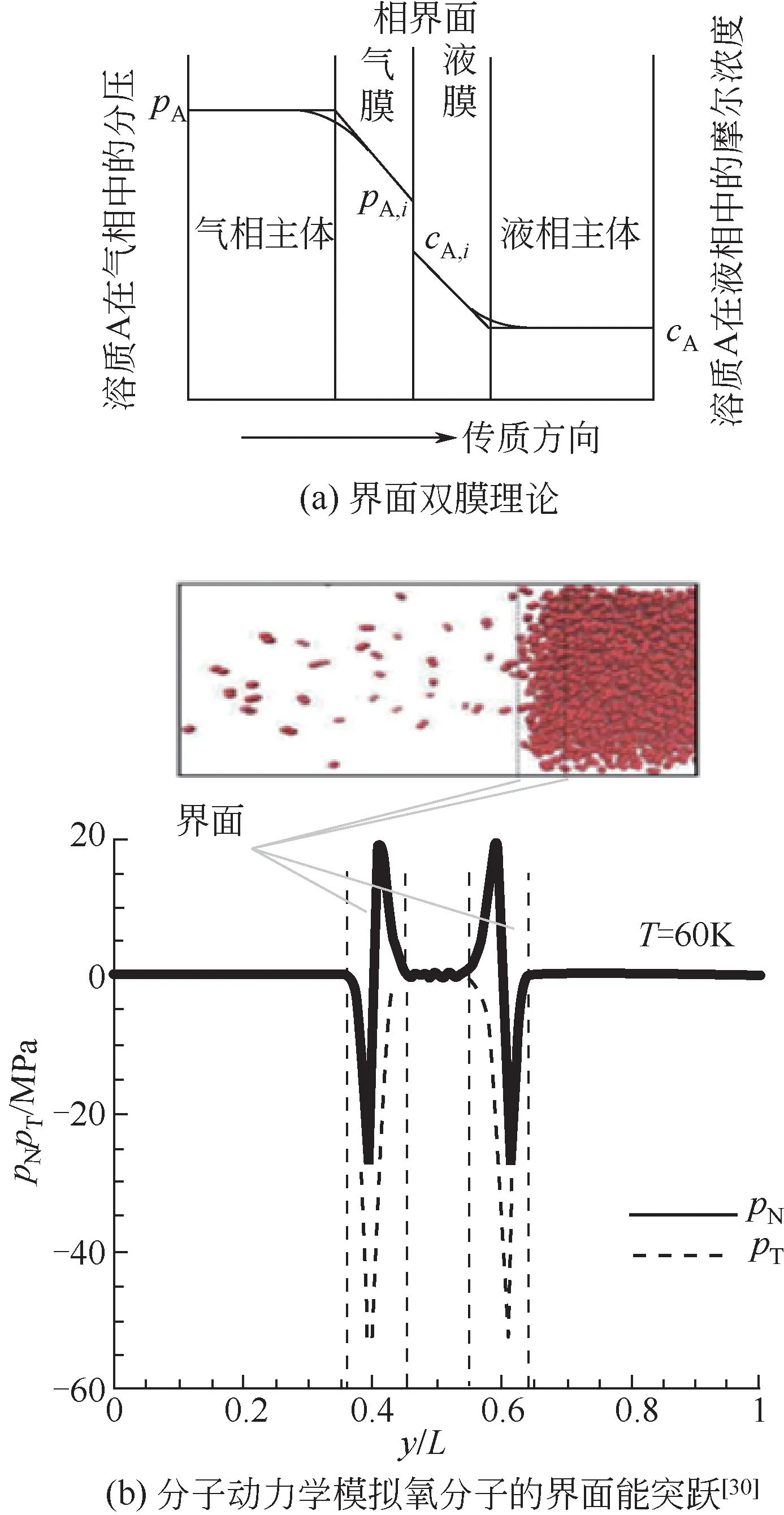
图6 界面双模理论及界面突跃
在微观界面能垒存在的基础上,分子需消耗能量进入界面区,因此在界面处同样存在界面内侧和外侧的微观差异,形成微观界面能差腐蚀。本文实验中气泡内含有氧气,气相分子的动能高于界面处分子的动能,O2分子由气相扩散进入液相时引起的能量损耗导致界面处能量突跃,如图6(b)所示,气液界面两侧的势差导致界面两侧形成较大的能量梯度,在界面处进一步叠加部分腐蚀速率,称其为微观界面能差腐蚀。气液界面的存在引起微观界面能差腐蚀及宏观氧浓差电池腐蚀,两者构成界面腐蚀,共同作用并增大铜试样的整体腐蚀速率。界面腐蚀的两个部分主要受浓度梯度即扩散控制的影响,使得浓度越高界面腐蚀越严重,在铜片上留下更清晰的界面腐蚀环线。
如图7(a)所示,铜电极表面带正电荷,溶液中的Cl-紧密地吸附在电极表面引起Cl-腐蚀,均匀液相腐蚀电流密度记为I1[32]。随Cl-浓度的升高,双电层电阻Rct逐渐减小,腐蚀电流逐渐增大;但伴随腐蚀产物CuCl1-mm 的积累,Cl-向电极表面的吸附和产物CuCl向溶液中的扩散受到制约,阻碍金属溶解过程的进行[33],Cl-吸附出现极限吸附值,此后腐蚀速率出现减小的趋势。如图7(b)所示,Cl-和O2分子在气-液-铜界面附近富集;界面外侧液相区域的腐蚀为阳极区域,界面内侧气相区域为阴极区域,形成宏观氧浓差腐蚀,腐蚀电流记为I2(A),有效腐蚀面积记为A2(cm2);气液界面层处的微观界面能差腐蚀反应电流密度记为I3(A),有效腐蚀面积记为A3(cm2);在忽略气泡内腐蚀速率的情况下,引入界面后的总腐蚀电流密度由液相腐蚀电流密度I1(A)和界面腐蚀电流密度I界面(A)两部分组成,且界面腐蚀电流I界面又可分为宏观氧浓差电池腐蚀电流I2和微观界面能差腐蚀电流I3,故结合考虑三部分腐蚀面积的影响,可得存在界面腐蚀时总腐蚀电流密度的表达见式(3)。
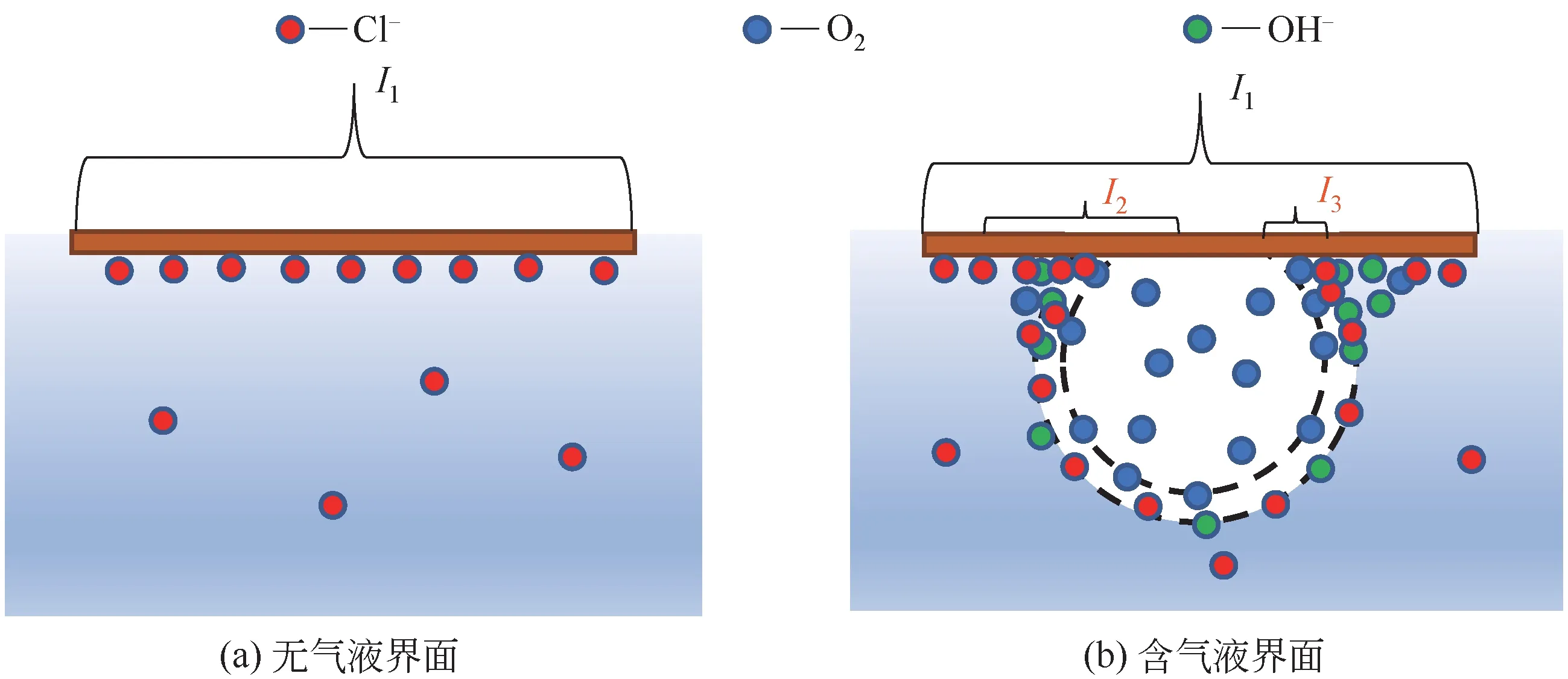
图7 溶液内界面处粒子分布示意图
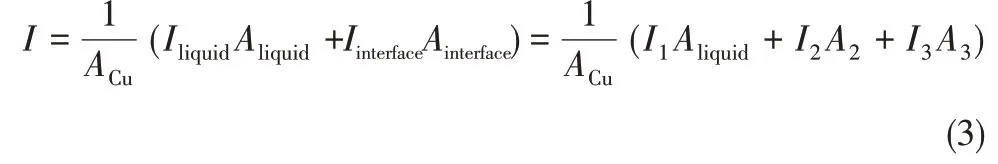
由式(3)可知只要存在界面I2和I3均不为0,界面的引入使腐蚀恶化。需注意的是,与无界面腐蚀中Cl-浓度存在浓度极值相类似,气相内O2含量有限,随反应的进行被快速消耗,界面处OH-离子浓度降低甚至供给不足,导致腐蚀电流密度I2和I3均减小;界面另一侧Cl-浓度影响同样存在浓度极限,两者共同作用使得界面腐蚀速率随浓度的变化呈现先增大后减小的变化规律。同时,通过本文机理及数据规律的分析,虽然不能将界面腐蚀所包含的宏观氧浓差电池腐蚀速率及微观界面能差腐蚀速率进一步进行分离,但两部分的叠加作用所引起的界面腐蚀速率Iinterface可根据式(3)计算从试样的总腐蚀速率中剥离。
3 结论
通过利用在液相中鼓入气泡的方法,获得稳定可控的气液界面,同时控制其他条件均相同的情况下对有无气液界面的铜试样进行对比实验研究,用实验和理论的方法获得含静态气液界面腐蚀规律,具体结论如下。
(2)金属Cu 表面静态气液界面的存在导致腐蚀电位升高、腐蚀电流增大;即界面的引入使试样较难发生腐蚀,但一旦腐蚀开始其腐蚀结果更为严重。
(3)由于界面腐蚀的基础是界面双膜理论,因此界面腐蚀速率主要受离子浓度的影响。有无界面工况下试样的总腐蚀速率均随Cl-浓度的升高先增大再减小,但剥离出的界面腐蚀电流随Cl-浓度的升高而增大。结合阻抗谱图高浓度下为扩散过程控制的腐蚀机理可知,界面腐蚀对总腐蚀速率的影响随浓度的升高而愈发显著。
