EIF2S3 基因变异致MEHMO 综合征2 例报告并文献复习
2021-03-29刘晓鸣王海侠谈倩倩
刘晓鸣 王海侠 陈 娇 张 园 谈倩倩
1.徐州市儿童医院神经内科(江苏徐州 221006);2.武汉康圣达医学检验所有限公司(湖北武汉 430075)
MEHMO 综合征患者以智力障碍、癫痫发作、生殖器发育不全、小头畸形和肥胖为主要特征。1989年首次被描述为一种综合征,1998年命名为MEHMO综合征[1-2]。MEHMO 综合征由X 染色体上EIF2S3基因变异引起。国外仅有20 余例MEHMO 综合征案例报道,其临床表型具有异质性[2-8],国内尚未见报道。MEHMO综合征临床表现的严重性可能与EIF2S3基因变异类型和定位所导致编码蛋白功能损害的程度有关[6]。本文回顾分析2 例MEHMO 综合征患儿的临床表型及遗传学特点,并进行文献复习,以提高对MEHMO综合征的认识。
1 临床资料
例1,男,G1P1,足月顺产,出生体质量3.2 kg,无产伤或窒息史。6 月龄竖头,1 岁始能主动翻身、偶能逗笑,1 岁4 个月能弓背屈腿坐、不能扶站。父母体健,智能无异常,无血缘关系。患儿1 岁3 个月始出现抽搐,当时体温38.5 ℃,表现为意识丧失,双眼凝视,面色发绀,流涎,四肢强直伴抖动,伴大便失禁,持续2~3 分钟,间歇期如常,后间隔数月有2 次体温超过38 ℃时抽搐,表现类似。2 岁2 月龄及3 月龄患儿各出现无热抽搐1 次,表现同前,持续2~3 分钟。患儿于2 岁3 月龄就诊于徐州市儿童医院。当时身高78 cm(-3 SD~-2 SD),体质量15 kg(1 SD~2 SD),头围44 cm(-3 SD~-2 SD);神志清,智力低下,小额,耳廓畸形,颈软,四肢肌张力增高,仅能扶站,不能独走,能追视,叫名反应慢,能逗笑出声,不能听懂简单指令,偶发简单的单字音。实验室检查:血乳酸5.8 mmol/L;血常规、肝肾功能、电解质、血糖、免疫功能及心肌酶等无异常。心电图无异常。头颅磁共振(MRI)示脑胼胝体薄、白质少(图1 A、1 B)。视频脑电图(VEEG)示清醒安静状态下枕区可见中低幅4~7 Hzθ波及中幅3~3.5 Hzδ波,左右欠同步对称,可见阵发性弥漫性δ活动,未观察到临床发作或癫痫样放电。予口服左乙拉西坦抗癫痫,患儿抽搐次数减少,未减停药物。2岁9月龄再次出现无热惊厥,复查血糖无异常,血乳酸3.9 mmol/L;VEEG示清醒安静状态下可见弥漫性不规则中低幅2.7~7 Hz混合慢波活动,双侧欠同步对称,睡眠期双侧前头部偶见尖波、尖慢波散发(图2A、2B)。加用丙戊酸钠后患儿无惊厥发生。随访至4岁4月龄,患儿身高95 cm(-3 SD~-2 SD),体质量17 kg(0 SD),头围46 cm(-3 SD),爱张嘴笑,易流口水,逐渐出现痉挛性肢体截瘫,不会说话,隐睾,随机检查生长激素4.13 ng/mL,生长激素激发试验(精氨酸和胰岛素)测生长激素峰值为12.01 ng/mL。
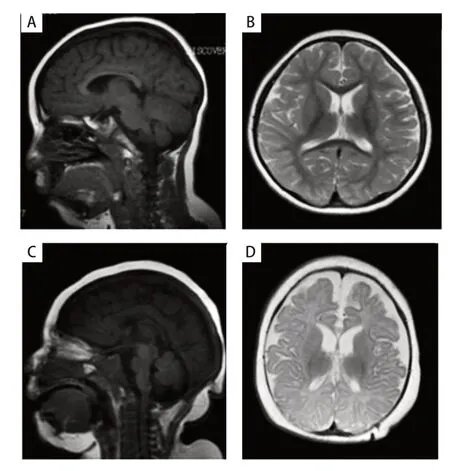
图1 患儿头颅MRI 表现
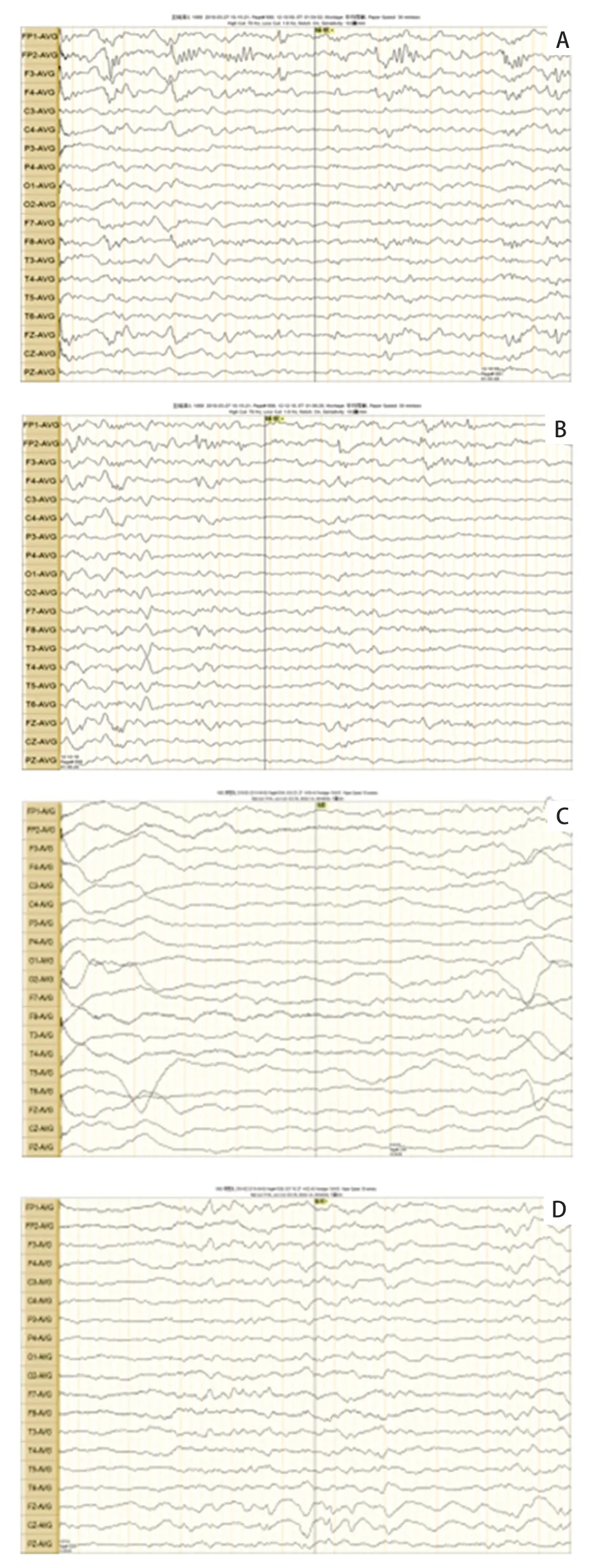
图2 患儿VEEG 表现
例2,男,G1P1,足月顺产,出生体质量3.6 kg。生后5 天后因少哭、少动伴呼吸急促转入新生儿科,诊断为新生儿缺氧缺血性脑病,蛛网膜下腔出血,新生儿肺炎,新生儿高胆红素血症,臂丛神经损伤,头皮血肿。父母体健,智能无异常,无血缘关系。入院体格检查:精神反应差,头围31 cm(-3 SD~-2 SD),前额小,两肺呼吸音粗,心脏无异常,阴茎短小,阴囊可及囊性包块,双侧睾丸鞘膜积液。实验室检查:血乳酸3.7 mmol/L,血糖1.7 mmol/L。予抗感染、蓝光照射、营养神经及对症治疗10天后好转出院。患儿3月龄时无明显诱因出现抽搐,表现为频繁眨眼,口唇发绀,无口吐白沫,四肢稍强直,无大小便失禁,持续约1分钟自行缓解,间断发作,每天或数天发作1、2次,未予特殊治疗。3月30日龄因反复抽搐再次入院。入院体格检查:头围33 cm(< -3 SD),小额,不能竖头,斜视,无注视及追视,双下肢肌张力稍高,阴茎短小。实验室检查:血乳酸3.9 mmol/L,其他生化检查无明显异常。头颅MRI示胼胝体较薄,双侧半卵园中心髓鞘化信号较少,内囊前肢髓鞘化信号未出现,双侧侧脑室后角增大,两侧额、顶、颞叶脑沟增深,邻近脑外间隙增宽,余脑室系统无扩大(图1C、1D)。VEEG示清醒状态各可见低幅2~8 Hz混合波,大致同步对称,余导联未见异常快波及慢波,睡眠期稍多量多灶性低-中波幅尖波、尖慢波散发(图2C、2D)。诊断为癫痫,小头畸形,小阴茎,全面发育落后。予奥卡西平治疗,患儿仍时有阵发性抽搐,易呈癫痫持续状态。家长要求出院,4月15日龄死亡。
因2 例患儿诊断不明予行遗传变异检测,包括家系全外显子组测序及拷贝数变异(copy number variations,CNV)检测。经医院医学伦理委员会审批[(2017)伦审第(1-20)号],患儿父母签署知情同意书,抽取患儿及其父母外周血各2 mL,置乙二胺四乙酸抗凝管中,用Takara Blood Genome DNA Exyration Kit试剂盒标准流程提取基因组DNA。全外显子组测序和CNV检测由武汉康圣达医学检验所有限公司完成。使用高通量测序(Illumina×10)技术进行全外显子组测序。测序片段通过Burrows Wheeler Aligner(BWA)软件(0.7.9a版)和UCSC hg 19人类参考基因组进行比对,比对结果采用Picard(1.115版)软件去除重复序列。应用基因检测智能操作系统进行变异注释和解读,结合ClinVar、在线人类孟德尔遗传(Online Mendelian Inheritance in Man,OMIM)和人类基因变异数据库(Human Gene Mutation Database,HGMD)等疾病数据库查找变异位点或疑似致病突变的收录情况,进行基因变异致病分析。SIFT、PolyPhen 等软件对错义变异进行致病性评估。根据全外显子组测序分析结果,使用Primer-BLAST 在线软件(https://www.ncbi.nih.gov/tools/primer-blast/)设计目标区域扩增引物。c.137C>T验证正向引物:5’-cca tgt tgg cca gga tggt-3’,反向引物:5’-TGG TCC CTG GAA TGT CCG TA-3’;c.1391_c.1394delCAAT验证正向引物:5’-ACC TAC TAA GGT AAC GAA CTG TGA-3’,反向引物:5’-TTC TTT ATG GAG GAG CAG GCT TTG-3’。以基因组DNA为模板,使用Taq DNA聚合酶扩增。PCR反应条件:95 ℃预变性5 min,95 ℃变性30 s,60 ℃退火30 s,72 ℃链延伸30 s,扩增30个循环;72 ℃补充延伸10 min。PCR体系均为50 μL。1%琼脂糖凝胶电泳检测PCR产物,纯化的PCR产物在全自动测序仪ABI377(Applied Biosystems,美国)上进行双向测序,测序结果用Chromas(2.23版)软件分析,并在NCBI中与正常序列(NM_001415)比对分析。
例1 患儿EIF2S3基因第3 号外显子存在1 处半合子变异,c.137 C>T 错义变异导致苏氨酸变为异亮氨酸(p.Thr46Ile)。此变异在HGMD等数据库未见报道。参考美国医学遗传学和基因组学学院(American College of Medical Genetics and Genomics,ACMG)基因变异解读指南,该位点为无家族史的新发变异(PS 2),位于变异热点和/或关键和既定的功能域的非良性变异位点(PM1),无人群携带率(PM2),SIFT及PolyPhen程序预测变异会导致基因产物的损伤效应(PP 3),与临床表型相符(PP 4)。ACMG 分级为致病(Pathogenic)。例2患儿EIF2S3基因第12号外显子存在1处半合子变异,c.1391_c.1394delCAAT缺失导致移码变异(p.Thr464Thrfs*5),使得氨基酸的编码提前终止。参考ACMG基因变异解读指南,该位点的LOF 变异导致基因功能可能丧失(PVS 1),与HGMD数据库中已确定的致病变异c.1394_1397delTCAA,p.(Ile465Serfs*4)有着相同的氨基酸改变(PS1),隐性遗传病MAF<0.005,属于低频变异(PM2),与临床表型相符(PP4)。ACMG分级为致病(Pathogenic)。
例1 的一代测序结果与全外显子组测序结果一致,为新发的错义变异,其父母均为野生型(图3)。例2 的一代测序结果与全外显子组测序结果一致,其半合子变异来自其母亲,母亲为杂合变异,父亲为野生型(图3)。两个位点变异均符合X染色体隐性遗传疾病发病机制,符合先证者及其家系成员表型及基因型的共分离特点。

图3 2 例患儿家系EIF2S3 基因突变一代测序结果
染色体全基因组芯片检测分析C N V。采用CytoScan 750 K 试剂盒(ThermoFisher Scientific,USA),按照试剂盒标准操作流程进行DNA消化反应、连接反应、PCR反应、PCR 产物纯化、标记反应、杂交反应、洗染、扫描等一系列试验过程,扫描得到的原始数据经过ChAS(version 4.1)软件分析,观察所有染色体的畸变。应用OMIM、Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources、Clinical Genome Resource、Database of Genomic Variants、Cytogenetics Array Group CNV database、The portal for rare diseases and orphan drug等公共数据库分析CNV 的收录情况,并评估其致病性。经染色体全基因组芯片检测分析,2 例患儿的染色体均未检测到致病性CNV改变。
2 讨论
以“EIF2S3基因变异”、“MEHMO 综合征”为关键词在中国知网、万方数据库及维普数据库收集中国MEHMO 综合征患者相关文献,以“EIF2S3gene mutation”、“MEHMO syndrome”为关键词在PubMed数据库中检索国外MEHMO 综合征患者相关文献。截止日期为2020 年4 月10 日。检索到临床资料和基因信息较全的MEHMO 综合征报道,英文文献5篇[4-8],未检索到中文文献。报道患者均来自国外,包含18 例患者,5 个EIF2S3基因变异位点:1 个移码变异c.1394_1397delTCAA(p.Ile465Serfs*4)和4个错义变异c.324T>A(p.Ser108Arg)、c.665T>C(p.Ile222Thr)、c.777T>G(p.Ile259Met)和c.1294C>T(p.Pro432Ser),均为半合子变异。移码变异患者表现出MEHMO 综合征全部重要表型(智力障碍、癫痫发作、生殖器发育不全、小头畸形和肥胖),不同错义变异患者的临床症状较轻,表现出部分表型。患者还表现出其他的临床表型,包括糖尿病、垂体机能减退、轴性低肌张力,四肢高肌张力、自闭症特征、面部畸形、肺心病等,患者间表型存在差异。多数患者的脑MRI显示薄胼胝体。
EIF2S3基因位于X 染色体,编码真核翻译起始因子-2(eIF2)的核心亚基eIF2γ。eIF2是一种异三聚体GTP 结合蛋白,参与合成Met-tRNA(i)向40 S核糖体的募集,eIF 2 复合物对蛋白质合成至关重要[5]。eIF 2 γ 蛋白包含3 个功能结构域(GTP-binding domain、Domain Ⅱ、Domain Ⅲ)。位于关键和/或既定的功能域的非良性突变位点可造成蛋白功能损失,引起疾病的发生。截止目前,HGMD 和ClinVar 等数据库及PubMed等收录文献仅报道了EIF2S3基因的6个变异位点,c.324T>A(p.Ser108Arg)、c.451G>C(p.Val151Leu)、c.665T>C(p.Ile222Thr)、c.777T>G(p.Ile259Met)、c.1394_1397delTCAA(p.Ile465fs*4)和c.1294C>T(p.Pro432Ser)[4-9]。
本组例1 的EIF2S3基因有1 个c.137 C>T(p.Thr 46 Ile)错义变异,例2 的EIF2S3基因有1 个1391_c.1394delCAAT(p.T464Tfs*5)移码变异。两个变异在HGMD 等数据库均未见报道,ACMG 分级均为致病。例2 的移码半合子变异来自其母亲(体健),外公、外婆、舅舅及其他亲属均体健,未行基因检测,但其母亲的杂合变异为新发的可能性大。结合临床表型和其他检查结果,2 例患儿均可诊断为MEHMO 综合征。
例1的错义变异位于eIF2γ蛋白的GTP-binding domain 功能结构域,临床表型为癫痫发作、智力低下、进行性痉挛性四肢瘫、小头畸形、面部畸形、身材矮小和隐睾,未见肥胖症。MRI 显示脑白质少、胼胝体薄,血糖、生长激素水平正常,乳酸水平稍高。错义变异c.324T>A(p.Ser108Arg)和c.665T>C(p.Ile222Thr)位于GTP-binding domain功能结构域,c.777T>G(p.Ile259Met)和c.1294C>T(p.Pro432Ser)错义突变分别位于Domain II和C-terminal domain功能结构域[4-6,8]。文献报道,不同功能结构域存在的错义突变导致的MEHMO 综合征患者之间临床表型不完全一致,但智力障碍、小头畸形、癫痫、生殖器发育不全和发育迟缓等为主要临床表型,头颅MRI 显示胼胝体薄等异常。实验室检查需考虑血糖、乳酸及生长激素水平的变化,分子遗传学分析可帮助疾病的诊断。
例2的c.1391_c.1394delCAAT(p.T464Tfs*5)移码变异和已报道的c.1394_1397delTCAA(p.Ile465Serfs*4)移码变异有着相同的氨基酸改变,导致C 端的氨基酸在469 位提前终止。临床主要表现为癫痫、小头畸形和小阴茎,存在新生儿低血糖和乳酸酸中毒。头颅MRI 胼胝体较薄及其他异常。患儿癫痫状态持续、难治,4 月龄死亡。同时例2 在出生后就表现出新生儿缺氧缺血性脑病、蛛网膜下腔出血、新生儿肺炎、新生儿高胆红素血症等症状。文献报道EIF2S3基因c.1394_1397delTCAA(p.Ile465Serfs*4)移码变异导致的MEHMO 综合征家系中,4 例男性患者表现出相似的临床表型,包括新生儿低血糖、严重脑积水、全面发育迟缓、小阴茎、身材矮小、癫痫发作和早期死亡[5]。随后报道该移码变异导致的MEHMO 综合征患者均表现出综合征的全部重要表型,且还表现出糖尿病和垂体功能减退等症状[6-7]。因此,C端移码变异可能导致eIF 2 γ 蛋白功能丧失,患者表现出MEHMO 综合征全部重要表型,且临床症状更严重,婴幼儿期死亡案例较多。另外,本组2 例患儿临床均表现出高血乳酸,这在之前西方人病例中未见报道,因此,血乳酸增高可能为东亚人MEHMO综合征表型之一。
采用酵母模型评估基因型-表型的关系发现,与移码变异导致患者更严重症状一致,eIF 2 γ-Ile 465 Serfs*4 变异显著损害酵母细胞生长,而表达eIF2γ-Asp167Arg和eIF2γ-val281hr变异的酵母(分别对应于人类eIF2γ中的Ser108Arg和Ile222r变异)与表达野生型eIF2γ的对照菌株酵母细胞生长水平相似,提示存在一定的基因型-表型关系[6]。C端移码变异导致患者表现出MEHMO 综合征的全部重要表型且表现严重,蛋白质功能结构域中的错义变异导致患者表现出部分表型且表现较轻,这需要更多的EIF2S3基因突变案例来明确。
综上,本研究首次报道中国两个MEHMO综合征家系,且全外显子组测序鉴定出两个新的变异,丰富了EIF2S3基因的变异谱,为明确MEHMO 综合征的基因型-表型关系提供了依据。
