基于分子对接结合靶标mRNA 表达的交泰丸镇静催眠药效物质及机制研究
2021-03-21韩洪翠张艳玲路芳毕磊黄运芳马鹏凯张玉杰
韩洪翠,张艳玲,路芳,毕磊,黄运芳,马鹏凯,张玉杰
北京中医药大学中药学院,北京 102488
交泰丸是治疗失眠症的经典名方,由黄连、肉桂按10∶1 组成,具有交通心肾、镇静安神功效,用于心肾不交引起的惊悸、夜寐不安等症。目前,交泰丸研究多集中于药理、化学成分和临床等,且镇静催眠作用是研究较多的方向之一[1],但由于交泰丸成分复杂,其镇静催眠药效物质及作用机制尚未完全明确。已知交泰丸可通过调节5-羟色胺(5-HT)、γ-氨基丁酸(GABAA)等中枢神经递质发挥镇静催眠作用[2-3]。因此,本实验采用分子对接技术,选取与中枢镇静催眠、降压等密切相关的3 个蛋白受体,即5-HT1A受体、GABAA 受体、多巴胺D3(DRD3)受体作为蛋白靶标,研究其与交泰丸主要成分小檗碱(BBR)、黄连碱(COP)、巴马汀(PAL)、表小檗碱(EBBR)、药根碱(JAT)的结合活性,同时结合蛋白mRNA 表达实验,探究交泰丸镇静催眠药效物质及其治疗失眠的分子作用机制。
1 实验材料
1.1 动物
SPF 级4 周龄雄性昆明小鼠56 只,体质量18~20 g,斯贝福(北京)实验动物科技有限公司提供,动物许可证号SCXK(京)2011-0004。饲养于北京中医药大学SPF 级环境,12 h 光照/黑夜循环,温度25 ℃,湿度60%,自由摄食饮水。
1.2 药物与试剂
BBR、PAL 对照品(批号分别为713-8702、732-8701),中国食品药品检定研究院,纯度≥98%;COP、EBBR、JAT 对照品(批号分别为MUST-12111602、MUST-14101309、MUST-14090711),成都曼思特生物技术有限公司,纯度≥98%;对氯苯丙胺酸(PCPA),批号20131101-17,美国Sigma 公司;Trizol Reagent试剂、反转录试剂盒、荧光定量PCR 试剂,Invitrogen公司;DEPC 水,北京鼎国昌盛生物技术有限责任公司;其他试剂均为分析纯。
1.3 仪器
Q-5000 型紫外分光光度计(Quwell 公司),WD-9413 型凝胶成像分析仪(北京六一仪器厂),DG-3D 型大型水平电泳槽、DG-Ⅲ型双稳数显电泳仪(北京东林昌盛生物科技有限责任公司),ABI PRISM 7500 型荧光定量PCR 仪(ABI 公司),3K18型离心机(美国Sigma公司),9600型普通PCR仪(ABI公司)。
2 实验方法
2.1 同源模建
5-HT1A受体同源模型多以2 种人β2 肾上腺素能受体[4]或牛视紫红质受体[5](PDB 代码分别为2RH1、3D4S、1U19,http://www.rcsb.org)为模板建立,将5-HT1A受体序列(P08908,http://www.uniprot.org)分别与以上3 种蛋白晶体结构进行序列对比,选取同源性较高蛋白为模板蛋白。用Discovery Studio2016 软件同源模建,得到20个模型,选择PDF Total Energy、DOPE score 均最低的模型作为目标蛋白。通过Ramachandran Plot 和Profile-3D 对同源模建蛋白模型进行评估。
2.2 分子对接
2.2.1 小分子配体及受体准备
本实验根据3 种受体蛋白的性质选择临床阳性药作为参考配体,见表1。

表1 靶标及参考配体信息
用ChemOffice2019 软件获得阿米替林、巴比妥、BP-897 和5 种黄连生物碱小分子初始3D 结构,用Discovery Studio2016 软件进行配体准备及能量最小化,得到优化后配体。导入同源模建得到5-HT1A受体蛋白,并在PDB 库中下载GABAA 和DRD3 受体的晶体结构,去掉结构中水分子和原配体,补充非完整的氨基酸残基,为蛋白加氢等,得到优化后受体。
2.2.2 活性位点确定与分子对接
根据5-HT1A受体与7 个配体对接结果[4],选择产生作用最多的ILE189 为活性氨基酸位点;GABAA受体α1 亚型(Gabra1)中PHE65 可决定配体亲和力,且相邻ARG67 可促进配体结合位点形成[6],因此,选择与ARG65 相邻PHE62 氨基酸残基为GABAA 受体活性结合位点;根据模建DRD3 活性氨基酸位点ASP117[7],选择位置相似、类型相同ASP110 氨基酸残基作为活性位点。基于以上活性氨基酸位点确定结合口袋,用Discovery Studio2016 软件Libdock 模块将3个受体分别与5 种黄连生物碱和对应的参考配体对接。
2.3 动物实验
2.3.1 造模及分组
小鼠适应性饲养1 周,随机分为对照组8 只和造模组48只。造模组腹腔注射PCPA(400mg/kg)混悬液,对照组注射等量生理盐水,连续2d,28~30 h观察小鼠行为[8]。造模组注射30h后白日活动量增加,无法入睡、摄食量减少、敏感性较强,表明造模成功。再分为模型组和5种黄连生物碱给药组各8只。
2.3.2 给药
造模第3 日,各给药组分别给予BBR、PAL、COP、EBBR 和JAT 5种黄连生物碱0.3g,分别用30mL生理盐水混匀灌胃,给药剂量100 mg/(kg.d),对照组和模型组给予等体积生理盐水灌胃,连续7 d。第7 日灌胃1h 后,断头处死,冰浴迅速取出全脑,液氮冷冻,置于-80℃冰箱保存。
2.3.3 qPCR 检测
Trizol提取小鼠脑组织总RNA。Q-5000紫外分光光度计测定其浓度及纯度,保证其纯度在1.7~2.0。取5μL RNA 溶液进行1%琼脂糖胶电泳,5 V/cm 电压电泳20 min,拍照,检测RNA完整性。然后以提取的总RNA 为模板,使用Invitrogen 反转录试剂盒进行cDNA的合成。PCR反应体系:cDNA 1μL,上游与下游引物各0.25μL,PCR 反应mix×(AmpliTaq Gold®FastDNA PolymeraseLD,10×buffer,SYBR®Green 1,dNTPs)12.5μL,灭菌水10.5μL,总反应体系25μL;反应条件:94℃预变性2min,94℃变性30 s,62℃退火30s,72℃延伸30s,共35次循环,72℃、10min 终止反应。采用2-ΔΔCt法对目的基因进行相对定量分析。引物均委托鼎国昌盛生物技术有限责任公司合成,引物序列见表2。

表2 PCR 各基因引物序列
3 结果
3.1 5-羟色胺1A 受体模型建立
5-HT1A受体序列对比结果显示,2RH1和序列的同源性与相似性最高,见表3。因此,以2RH1为模板进行同源模建,选择可信度与质量最高的模型为目标蛋白(见图1)。Ramachandran 图评估结果(见图2)显示,psi-phi 构象不合理氨基酸(红色点)有4个,小于氨基酸总数5%,表明5-HT1A受体模型氨基酸二面角结构较合理。Profile-3D评估结果显示,模型Verify Score、Verify Expected Low Score和Verify Expected HighScore分别为108.59、190.252、85.6135,且Verify Score位于Verify Expected Low Score和Verify Expected High Score 之间,说明模型质量较高。
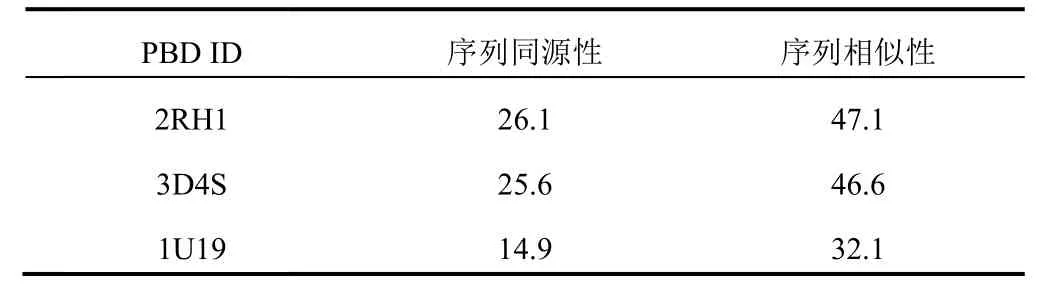
表3 5-HT1A 受体序列对比结果(%)
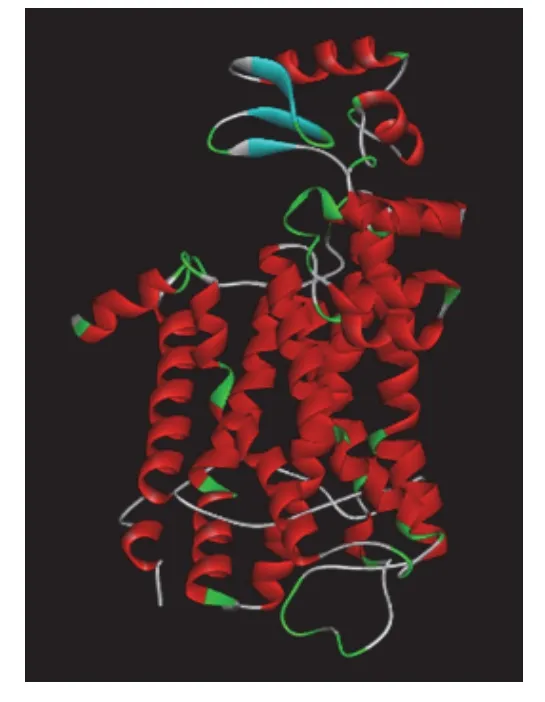
图1 5-HT1A 受体3D模型
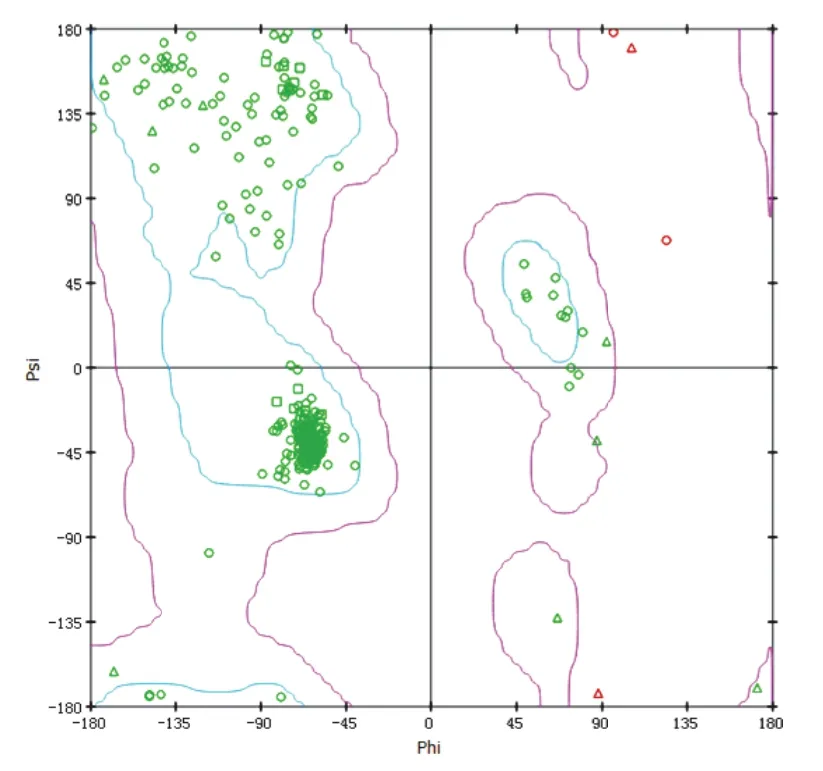
图2 5-HT1A 受体Ramachandran图
3.2 分子对接结果
LibDock分子对接结果见表4。5种黄连生物碱与5-HT1A、GABAA、DRD3受体打分结果均较高,表明配体小分子与受体结合活性均较高,且其中EB BR 打分高于参考配体阿米替林,5种生物碱打分结果均高于参考配体巴比妥,显示中药小分子针对受体蛋白靶向治疗失眠的潜力,而参考配体BP-897作为DRD3受体激动剂,其打分结果高于5种黄连生物碱,表现了BP-897对DRD3受体的高亲和力。配体小分子与5-HT1A、GABAA、DRD3受体蛋白的分子对接二维平面图(见图3~图5)显示,各对接构象同受体蛋白氨基酸残基之间相互作用力。经统计,配体主要与5-HT1A受体氨基酸残基GLY237、THR236形成氢键,加强小分子与蛋白的结合,同时与ASP239和ARG227等氨基酸残基产生静电作用和疏水作用;配体主要与GABAA 受体氨基酸残基LYS66、ALA72等产生疏水作用,与氨基酸残基LEU70、LYS66等产生氢键作用,极少产生静电作用;配体与DRD3受体蛋白之间产生的非键相互作用主要为氢键作用和疏水作用,产生作用的主要氨基酸残基分别为SER192、VAL111和PHE346,只与氨基酸残基ASP110产生静电作用。

表4 LibDock 分子对接结果(分)
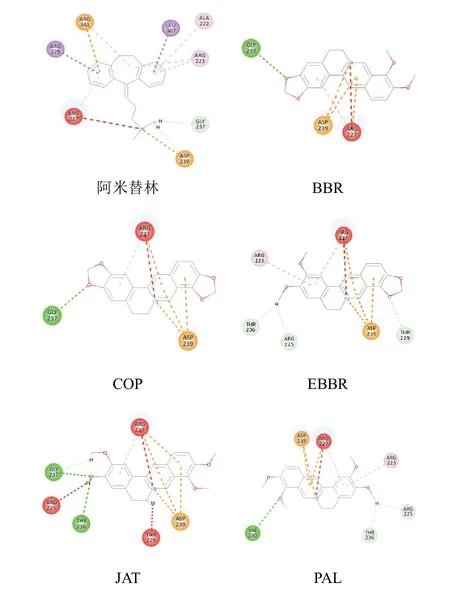
图3 各配体小分子与5-HT1A 受体相互作用二维平面图

图4 各配体小分子与GABAA受体相互作用二维平面图

图5 各配体小分子与DRD3受体相互作用二维平面图
3.3 qPCR 检测结果
与对照组比较,模型组小鼠脑组织5-HT1A、Gabra1和DRD3mRNA 表达显著下调(P<0.01);与模型组比较,COP组小鼠脑组织除DRD3mRNA表达上调不明显外,5-HT1A、Gabra1mRNA 表达显著上调(P<0.05,P<0.01),余4种生物碱组5-HT1A、Gabra1、DRD3mRNA 表达显著上调(P<0.05,P<0.01)。见表5。
表5 各组小鼠脑组织5-HT1A、Gabra1 和DRD3 mRNA 表达比较()

表5 各组小鼠脑组织5-HT1A、Gabra1 和DRD3 mRNA 表达比较()
注:与对照组比较,**P<0.01;与模型组比较,#P<0.05,##P<0.01
4 讨论
失眠症属中医学“不寐”范畴,是由脏腑机能紊乱、气血亏虚、阴阳失调引起的,导致不能获得正常睡眠的一类病症。交泰丸是治疗心肾不交型失眠的常用基础方剂,前期研究发现,交泰丸透过血脑屏障的主要成分为黄连生物碱,且其在病理动物模型上表现出显著差异的脑分布特征[9]。由此推测,黄连生物碱可能是交泰丸镇静催眠的药效物质。
当今,计算机辅助药物设计(CADD)、分子烙印技术等已成为药物研发领域的一部分[10]。而分子对接技术作为CADD 受体的一种方法,可迅速且较精确地预测受体和配体结合模式,在研究中药有效成分的潜在靶标及作用机制方面有独特的优势[11]。近年随着医学研究的推进,发现多数中枢神经递质在调节睡眠-觉醒节律中发挥了重要的作用[12]。研究表明,交泰丸催眠的药理作用与其影响调节睡眠中枢神经递质肾上腺素和5-HT 释放有关[2];交泰丸可增加PCPA模型大鼠GABAA 受体表达,从而发挥镇静催眠作用[3];此外,作为中枢神经系统重要的儿茶酚胺类神经递质,多巴胺可通过其特异性受体,发挥影响学习记忆、调节“睡眠-觉醒”、参与认知与情感等多种复杂的生理功能,在多巴胺受体中D3 受体亲和力最高。因此,本实验选择5-HT1A、GABAA、DRD3 受体做为分子对接靶标,与交泰丸的5 种主要黄连生物碱及相应的参考配体进行分子对接打分,预测配体与受体之间的结合活性。结果显示,5-HT1A、GABAA、DRD3受体与配体之间打分结果均较高,结合5 种黄连生物碱与受体之间的结合活性,可看出结合活性较高的原小檗碱型生物碱为EBBR、BBR 和COP。
为进一步探究交泰丸镇静催眠的药效物质及其分子机制,本研究使用PCPA 建立失眠小鼠模型,通过测定小鼠脑组织5-HT1A、Gabra1 和DRD3 受体mRNA 的表达,评估交泰丸镇静催眠药效物质对中枢神经递质受体的影响。结果显示,除COP 组DRD3受体表达上调不明显外,5 种黄连生物碱均可显著上调5-HT1A、Gabra1 和DRD3 受体表达,其中上调作用较明显的生物碱为BBR、PAL 和JAT。结合分子对接与靶标蛋白mRNA 表达实验结果显示,小檗碱结合活性与分子作用强度均较高,而结合活性较高的EBBR 与COP 分子作用强度不高。造成此差异的原因可能是5 种黄连生物碱成分在生物体内跨血脑屏障转运能力不同,由于小分子化合物与血浆蛋白结合后不易透过血脑屏障,结合黄连生物碱进入脑中的含量差异[13]与其血浆蛋白结合率的差异[14],可以看出BBR跨血脑屏障运输的能力强,而EBBR 与COP 不易透过血脑屏障;此外,3 种神经递质受体在小鼠脑组织分布差异也可能是导致2 个实验结果差异的原因之一。在交泰丸分子作用机制方面,5 种黄连生物碱对中枢神经递质受体的影响结果表明,原小檗碱型生物碱能提高5-HT1A、Gabra1、DRD3 受体mRNA 的表达,从而增加神经递质5-HT、GABAA、多巴胺与受体的结合,促进慢波睡眠形成[12],抑制啮齿动物本能运动[15],起到治疗失眠作用。
本研究选择已经证实的与中枢神经系统镇静催眠、降压等作用密切相关的3 种受体探究交泰丸镇静催眠药效物质及其治疗失眠的机制,表明原小檗碱型生物碱成分是交泰丸的镇静催眠药效物质,其中小檗碱作用最强,且可通过影响5-HT1A、Gabra1、DRD3等受体的表达治疗失眠,但由于黄连生物碱具有多靶点的特性,其机制有待进一步研究。
