肝硬化PHT症发病机制研究进展
2021-03-21李奇素刘永刚南然惠建萍李京涛魏海梁闫曙光
李奇素,刘永刚,南然,惠建萍,李京涛,魏海梁,闫曙光
【提要】 门静脉高压是肝硬化并发症之一,其发生机制较为复杂,肝内梗阻是最为直观的病理机制,肝内梗阻分为机械梗阻和动态成分两种因素,分别通过不同的机制影响门静脉压力。内脏血管扩张通过增加门脉血流量而加重门静脉高压,而内脏血管扩张基于血管扩张物质的释放、血管收缩剂反应性降低和神经相关因素导致PHT。本文对引起PHT的肝内外因素进行综述,将下一步研究和临床治疗广泛结合提供新的研究思路,启发新的治疗靶点,以期为该领域的研究奠定理论基础。
肝硬化是引起门静脉高压及其并发症最常见的原因,持续的肝脏损伤导致纤维化和门静脉高压增加,当门静脉压力压力梯度(HVPG)>10 mmHg时可能出现失代偿和食管静脉曲张,在代偿性肝硬化中,食管静脉曲张的发生率分别约为每年7%~8%,且静脉曲张出现后,其直径以相似的速度增长,并且每年可能在5%~15%的患者中破裂出血[1]。全球每年约有200万人死于慢性肝病,其中100万人死于肝硬化并发症[2]。PHT症(portal hypertension,PHT)是由于肝内循环的解剖和功能变化导致的门静脉对输入静脉流量的阻力增加以及内脏血管扩张和心输出量增加导致的门静脉流量增加的累积效应[3]。肝硬化假小叶形成、肝内机械梗阻和肝内皮细胞功能障碍、肝星状细胞活化等动态成分引起肝脏血管生成,门静脉血流受阻形成PHT继发侧支循环,外周血管扩张进一步引起全身血液高动力循环,加重门静脉高压。因此,探索PHT的发病机制对该病潜在治疗靶点具有重要意义。
1 肝血管梗阻
肝血管阻力增加是由肝结构改变引起的肝内梗阻和肝血管张力改变共同作用的结果,20世纪80年代以前使用“后向血流”学说来阐述肝硬化门静脉高压的形成机制,即硬化肝脏内的假小叶以及血管紊乱重构共同促使门静脉阻力增加,导致门静脉血流减少、内脏血流淤滞和低灌注,最终形成PHT。PHT的外科门腔静脉分流术即是使用支架在肝静脉和门静脉系统之间创建了肝内分流,使之前受阻的血流绕过硬化的肝脏,缓解了PHT[4]。其中TIPS(经颈静脉门体肝内分流术)可有效缓解PHT,降低胃肠道黏膜瘀血程度,有报道指出,经TIPS术后患者短期内肝功能减退,可能是由于术后肝脏一过性缺血导致,随着时间推移,术后高动力循环解除,肠肝循环中炎症介质下降,使肝功能滤过及清除能力可逐渐好转[5]。
2 动态成分
肝硬化门静脉血流的肝阻力增加还伴有肝血管张力增加而产生的动态成分[6]。这种动态成分的原因来自肝窦内皮细胞(LESCs)的内皮功能障碍、活化的肝星状细胞(HSC)、肝内血管生成、血栓形成和其他血管收缩剂的作用。HSC是一种肝脏特异性间充质细胞,在正常肝脏中,HSC保持静止(QHSCs),含有维生素A和大量脂滴;在肝损伤时,从受损的肝细胞和免疫细胞接收自分泌或旁分泌信号转变为高度激活、增殖、运动和收缩的肌成纤维细胞表型,称为肝星状细胞激活(AHSCs),具有调节内皮增殖、收缩调节血管张力、细胞外基质的形成以及调节的功能[7]。
2.1 星状细胞(HSC)作用机制
2.1.1 内皮素1(ET-1) 在受损的肝脏中,HSC以自分泌和旁分泌的方式表达或应答ET-1。ET-1是一种有效的内源性血管收缩剂,通过G蛋白偶联受体(GPCR)发挥作用,ET-1的高亲和力发挥血管的收缩效应。HSC的活化增加了ET-1的产生,增强血管收缩和PHT[8]。HSC与成为ET-1的自分泌靶点,内皮素与G蛋白偶联受体内皮素A(ETA)和内皮素B(ETB)结合,这两种受体分别出现在血管平滑肌细胞(VSMC)和内皮细胞上,协同发挥血管收缩的作用,从而增加肝血管的阻力和张力[9]。在肝硬化动物模型中,ET-1已被证明旁分泌和自分泌因子来激活HSC并增加肝脏门静脉血流的压力,从而引起门静脉压力升高[10]。同时,在慢性肝损伤时,肝细胞分泌TGF-β、TNF-α、血小板活化因子(PAF)等多种促炎标志物,进而刺激LSECs分泌ET-1。研究表明TGF-β1是强大的致炎因子,对HSC有极强的活化作用,TGF-β1能够诱导细胞活化,加快肝纤维化的进程[11]。因此,PHT与HSC的激活和ET-1有着巨大的联系。
2.1.2 收缩调节血管张力 肝损伤时活化的HSC获得增殖、收缩和肌成纤维细胞(MFBs)样表型,活化的MFBs样表型分泌几种促纤维化的细胞因子和趋化因子以维持受损肝脏的病理生理状态,活化的HSC获得肌成纤维细胞表型并引起收缩[7]。肌成纤维细胞的收缩通常肌成纤维细胞由肌球蛋白轻链磷酸酶(MLCP)调节从而控制血管张力,该机制主要由G蛋白偶联受体介导,由肌球蛋白轻链激酶(MLCK)诱导的短期的、强烈的和钙依赖性的收缩,HSC的收缩使肝窦变窄,从而进一步增加肝脏阻力,导致和加重PHT[12]。
2.1.3 细胞外基质 肝纤维化的发展以细胞外基质(ECM)的过度沉积为基本特征,活化的HSC是纤维化肝脏中细胞外基质沉积的主要来源。纤维细胞起源于HSC,HSCs上调α-平滑肌肌动蛋白和其他肌成纤维细胞内微丝的表达,迁移到损伤部位并分泌ECM产生纤维瘢痕。纤维细胞除了分泌I型胶原、纤连蛋白和波形蛋白外,还表达和分泌TGFβ促进ECM沉积[13]。ECM在肝窦周隙的沉积不仅阻碍肝脏的代谢功能,而且与HSC的增殖和迁移相互作用,从而加重纤维化[14],这一过程进一步提高了血管张力,增加肝脏的硬度[8],肝硬化本身可能进一步促进HSC的激活,激活的HSC可能会由于HSC的趋向性而迁移到这些纤维化点,加重纤维化,形成肝硬化-PHT之间的恶性循环。
2.2 肝窦内皮细胞(LSECs)作用机制
动态成分除HSC的改变外,还包括肝窦内皮细胞(LSECs)结构和功能的改变,即肝窦内皮细胞毛细血管化和功能障碍两个方面。
2.2.1 毛细血管化 毛细血管化是指基底膜和其他基质蛋白的沉积,在早期纤维化过程中发生的LSECs结构的变化,由于肝纤维化和肝损伤,在血管内皮生长因子(VEGF)调控下,内皮细胞窗孔直径变小、数量减少,细胞间隙变窄、消失和基质蛋白沉淀,形成基底膜,使肝窦毛细血管化,最终导致肝窦到中心静脉的开口消失,且影响细胞内物质的转运,阻碍血细胞的通过,导致血液流通阻力加大,门静脉压力增高[15]。
2.2.2 内皮细胞功能改变 内皮细胞功能障碍导致血管舒张因子一氧化氮(NO)释放减少,缩血管因子ET-1分泌增多,血管平衡稳态被打破[16]。LSECs通过释放血管活性分子,在调节肝血管张力中发挥重要作用。在肝硬化和门静脉高压症中,LSECs失去了血管舒张特性,减少了血管扩张分子(如NO)的生物利用度,增加了血管收缩分子的水平,从而增加了肝内阻力[6]。
HSC和LSECs之间的联系也发挥了重要的作用,主要体现在以下几个方面:①LSECs衍生的外体中的脂质酶鞘氨醇激酶1(SK1)及其产物鞘氨醇-1-磷酸(S1P)是作用于HSC迁移的关键介质,为HSC的迁移提供了重要介质,而HSC迁移是激活的一个重要前提[17],为以后HSC的激活创造了条件;②由LSECs产生的一种细胞纤维连接蛋白含有选择性剪接的额外结构域A(EIIIA),通过此额外结构区域可以促进HSC的运动,与HSC以往的运动方式不同的是此结构不能分化成肌成纤维细胞来促进HSC的运动[18];③HSC和LSECs通过不同的方式影响ET-1,ET-1通过肝内微循环中的ET-1受体介导血管舒张和收缩。ETA位于HSCs和平滑肌细胞(VSMC)上,介导血管收缩。ETB根据其细胞位置的不同,既能引起血管收缩,又能引起血管舒张。ETB1受体驻留在LSECs上,通过NO的产生引起血管扩张,两种细胞通过不同的方式作用于ET-1,引起PHT[19]。肝内梗阻的动态成分机制主要包括HSC和 LSECs作用机制(图1)。
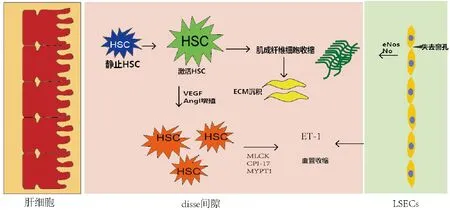
图1 动态成分对PH的作用机制
3 血管生成结构变化
肝内循环的血管结构变化包括血管生成和肝窦重构。血管生成是指内皮细胞(ECs)在先前的非血管组织中原位分化和增殖,然后结合形成原始的管状网络[20]。虽然LSECs是参与血管生成的最主要的细胞类型,但研究表明,HSC可以影响血管生成,这可能通过直接和间接机制发生[21]。直接机制包括HSC稳定新血管的能力,并为血管提供耐久性;间接机制包括HSC在缺氧时被激活,并通过增加VEGF、血管生成素-1(Ang1)及其相关受体的表达以招募和刺激LSECs,形成的新血管并确保其强度。HSC和LSECs之间的相互作用引起肝内血管生成和纤维组织沉积之间的恶性循环。LSECs产生血小板衍生生长因子(PDGF)和TGF-β1,促进HSC的募集和迁移[22]。
在肝脏中,血管生成可以发生在生理和病理状态下,生理状态下表现为肝再生。有实验研究证明人类活体肝移植(LDLT)中移植供体移植物的再生,显示血管和增殖内皮密度高,提示体内血管生成,且血管生成与供体移植体积成反比[23]。病理状态下表现为肝硬化和肿瘤血管生成,病理性血管生成是癌症和各种缺血性和炎性疾病的标志,其中缺氧和炎症是血管生成和血管重塑的强刺激因素[24]。微观上,持续炎症通过内皮间连接的松弛促进血管通透性,外渗的血浆蛋白和细胞外基质成分构成了内皮细胞迁移的支架[25]。新形成的血管响应肝硬化时肝脏形态功能重排而绕过窦道,不能向组织提供氧气和营养,这恶化了疾病的进程并增加了肝血管对门静脉血流的阻力[26]。新生血管形成在肝血管重塑、门静脉侧支循环形成、肠系膜充血、炎症介导的内脏血管扩张和高灌注中起着关键作用。HSC和VEGF呈相互促成的关系,HSC表达VEGF,反过来VEGF以旁分泌和自分泌的方式作用于相邻的LSECs和HSCs本身,进一步加剧了肝纤维化[27]。VEGF驱动生成新生血管,促进了内脏血流量的增加,同时促进形成门体侧支循环,与内脏血管扩张协同作用。
在PHT中,缺氧、炎症、剪切力和氧化应激的增加可能共同作用诱导VEGF的激活和病理性新生血管[28]。VEGF在新生血管形成的初始阶段起主导作用,激活内皮细胞的增殖,进而形成内皮小管。然而,新生血管的成熟主要受PDGF调节,它调节内皮小管与壁细胞和外周细胞的结合,从而稳定新生血管的构筑[24]。此外,胎盘生长因子(PIGF)也影响血管生成,PIGF表达血管内皮生长因子受体1(VEGFR1)的平滑肌细胞和成纤维细胞,也可能通过激活的内皮细胞释放细胞因子间接影响其增殖和迁移。通过这些作用,PIGF招募新生血管周围的平滑肌细胞,从而将它们稳定成成熟、耐用和不渗漏的血管[26]。在肝硬化肝脏中,炎症可能是连接血管生成的通路,当炎症细胞侵袭肝脏时,巨噬细胞、单核细胞、血小板和肥大细胞在趋化因子的介导下被招募到炎症区域,导致血管内皮细胞的增殖和迁移,形成新生血管[20]。
4 高动力循环状态
随着门静脉压力的增加,有两种主要的血管改变:一种是动脉扩张,进一步形成内脏高动力循环;另一种是门体侧支血管的形成。内脏动脉血管扩张是高动力循环的一个关键特征,使流入门静脉系统的血液持续增加,从而加剧门静脉高压[29]。门静脉高压症的高动力循环状态的本质是内脏和外周血管扩张、血浆容量增加和心输出量增加[30]。内脏高动力循环的原因(图2)也是多样的,主要包括血管扩张剂增多、血管收缩剂反应性降低、神经因素等。
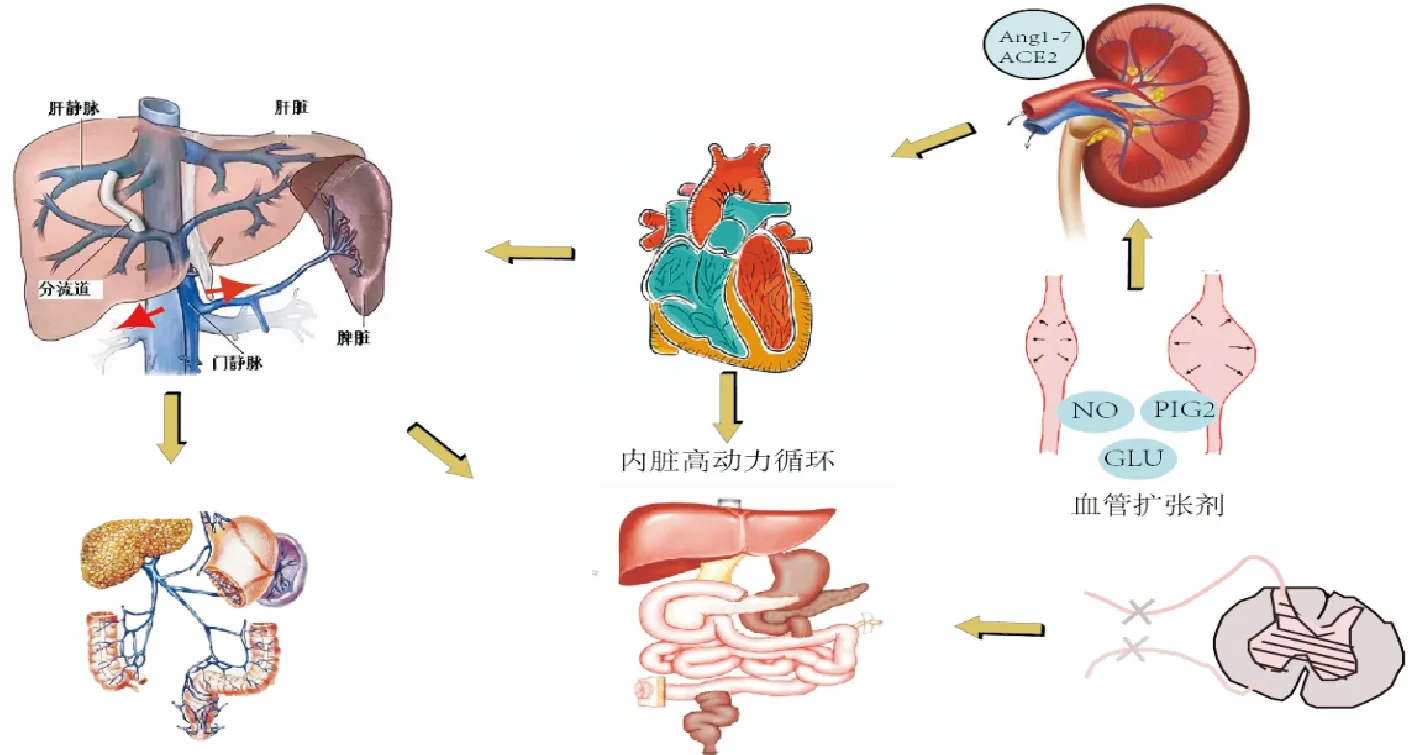
图2 内脏高动力循环与PHT发病机制
4.1 血管舒张物质
门静脉高压症的发生是因为血管阻力增加,随着PHT的发展,内脏血管扩张导致门脉血流量增加也是PHT的机制之一。在内脏血管中,与血管内皮功能障碍的血管扩张剂相反,是因为血管扩张剂的局部分泌促进血管扩张,固有的血管收缩能力降低引起血管扩张。
4.1.1 一氧化氮 一氧化氮(NO)可能是最重要的血管扩张分子,NO的产生有三种亚型:诱导型NO合成酶(INOS)、内皮型NO合成酶(ENOS)、神经元型NO合成酶(NNOS),结构性eNOS与不同的内皮细胞刺激,如剪切应力、炎性细胞因子和VEGF,可能刺激NO信号通路激活,增加NO合酶活性(eNOs)和NO合成,引起血管扩张[31]。NO引起血管舒张的机制是通过刺激可溶性鸟苷酸环化酶(sGC)在血管平滑肌中产生环磷酸鸟苷(CGMP),钾离子外流,并激活降钙素基因相关肽依赖的蛋白激酶,使肌球蛋白轻链激酶去磷酸化,导致血管舒张[32]。还有一种说法是NO产生的机制是细胞因子的介导,肝硬化腹水患者或动物有大量细菌穿过肠道屏障导致细菌副产物系统性释放,引发全身性内毒素血症,这可能刺激天然免疫细胞产生包括TNF-α在内的细胞因子,并通过刺激eNOS活性和刺激NO的产生[30],NO的产生加剧了内脏血管扩张,引起内脏血管的高流量状态。
4.1.2 前列环素 前列环素(prostacyclin,PGI2)是前列腺素(prostaglandin,PG)家族的重要成员,是一种内源性血管活性物质,是由环氧合酶(COX)在体液和物理刺激作用下合成的,导致PHT的内脏血管扩张和血管收缩减退。研究证实了PGI2的增加在实验性肝硬化门静脉压力升高和高动力循环发展中起作用,肝硬化大鼠和门静脉结扎大鼠的血浆和尿中PGI2水平高于对照组[32]。还有研究发现[33],NO与PGI2呈协同作用通过激活腺苷酸环化酶和增加环磷酸腺苷(cAMP)的产生,使邻近的血管平滑肌细胞松弛,导致PHT的内脏血管扩张和血管收缩减退。若PGI2水平的增加先于血流动力学变化的发展,这表明高动力循环主要是内脏血管内PGI2释放增加的结果。
4.1.3 胰高血糖素 胰高血糖素(GLU)是一种由胰腺α细胞释放的肽类激素,除了通过促进糖异生和糖原分解来调节葡萄糖水平外,还可以降低血管阻力。在肝硬化病理情况下,由于肝脏受损和大量侧支循环形成,GLU可以避免肝脏的降解。在门静脉高压症中,胰高血糖素导致高动力循环,以及肠系膜动脉对循环血管收缩剂反应性的降低[32]。研究发现[34]门静脉高压症患者的GLU水平升高,血清GLU水平与高全身血管阻力指数显著相关,GLU显著增加门静脉压力,并引起内脏血管扩张。且GLU显著增加门静脉结扎大鼠的门静脉压力,但不改变假手术大鼠的门静脉压力。便可以推测GLU引起的血管扩张只针对出现门静脉高压的病理情况下,而不针对正常的门静脉压力,表明GLU是PHT的结果,而不是导致的直接原因。
4.2 血管收缩剂反应性降低
在肾素-血管紧张素-醛固酮系统(RASS)中分为经典轴和交替轴,经典轴的成分血管紧张素转换酶(ACEI)和血管紧张素Ⅱ(AngⅡ)在肝内循环中显著增加,导致了PHT和肝内血管阻力增加。交替轴的成分包括血管紧张素转换酶2(ACE2)和效应肽血管紧张素(1-7)[Ang(1-7)]通过假定的受体Mas(MasR)介导发挥作用[35]。这些细胞过度表达血管紧张素Ⅱ受体-1(AT1R),并对升高的AngⅡ产生反应而收缩,AngⅡ有助于血管内皮细胞收缩,从而增加肝内血流阻力[36]。ACEI2、Ang(1-7)在肝硬化动物内脏血管床中的表达增加,实验表明给肝硬化大鼠输注Ang(1-7)降低内脏血管阻力的程度比对照组大得多,这表明Ang(1-7)是肝硬化内脏血管扩张的关键介质,这种血管活性途径在内脏器官中上调,但在肝硬化患者和大鼠的肝脏中也上调,在实验性肝硬化中,Mas受体激动剂抑制肝内血管收缩,减少肝纤维化和降低门静脉压[28]。肝硬化内脏血管中Ang(1-7)的血管舒张作用似乎是通过其受体MasR介导的。在肝硬化大鼠模型中,特异性MasR阻断剂A779增加了内脏血管的阻力,减少了内脏血流量,从而改善了门静脉高压,然而,通过阻断Ang(1-7)介导的肝脏血管扩张会增加肝内阻力,这一方法可能会损害MasR阻断通过增加内脏阻力来降低门静脉压力的有效性[35]。因此交替轴分泌的物质有助于介导血管扩张促进PHT。尽管传统RASS系统激活,血管紧张素Ⅱ(AngⅡ)分泌增多,血管收缩剂水平升高,但内脏血管扩张的高动力循环状态仍然存在,是因为内脏血管床对血管收缩剂的血管低反应性有关。无论是RASS的传统轴还是交替轴,各自介导的成分都会导致扩血管物质增加和减低内脏血管床对血管紧张素的敏感性,导致PHT的出现和进展。尽管RASS在肝硬化早期增加肝内阻力中起重要作用,但RASS在晚期肝硬化中增加肝内血管张力的作用可能被其他强大的血管收缩系统如内皮素和或交感神经系统的激活所抵消[36]。
4.3 神经机制因素
有研究发现肝硬化患者内脏动脉对交感神经刺激的压力反应明显减弱,部分原因是去甲肾上腺素(NA)释放改变、神经肽(NPY)释放不足,以及外源性NPY可显著恢复肝硬化大鼠的脱敏和血管收缩功能[37]。内脏血管张力的神经调节通过中枢和外周传出和传入神经系统。有实验证明,门静脉高压症时肠系膜血管的肾上腺素能改变和交感神经萎缩是由于神经调节的改变导致神经节后交感神经退行性变和细胞凋亡,从而导致内脏血管舒张[38]。
5 展望
肝硬化PHT症发病机制比较复杂,首先,肝内梗阻和内脏高动力循环两个因素中有一对矛盾,LESCs功能障碍引起的血管扩张剂兴奋性降低,这两因素均可以引起血管张力和血管阻力增大;而内脏高动力循环与LESCs功能障碍功能相反,引起血管扩张剂生成和血管收缩能力降低引起血管扩张;考虑到肝外和肝内机制的相反调节,笔者认为今后的研究方向可以从针对肝内外血管特异性着手,提高血管扩张肝内外的特异性,调控PHT,使肝内血管生成和内脏高动力形成一个良性循环。VEGF驱动生成新生血管,促进了内脏血流量的增加,形成内脏高动力,因此可以考虑研究抗VEGF生成,减轻门静脉压力,但是也存在一个问题,抗VEGF生成会阻断VEGF本来的活性,阻断VEGF生理性的血管生成作用,因此,仅阻断病理性血管生成不乏为改善PHT的主要治疗措施。PHT治疗缺乏良好治疗措施的原因可能是肝内分子病理的复杂性,其中HSC、LSECs各自介导的机制影响血管活性之外,其二者还能相互影响,现代研究虽然在分子层面已经逐渐深入了,但是还是缺乏系统性的靶向治疗,比如,以药物为载体单独针对HSC、LSECs细胞功能的研究。最后,RASS系统有传统轴和交替轴的提出,治疗上传统的RASS轴得以临床应用,交替轴可能比传统RASS轴更具有特异性的抑制内脏血管扩张,减慢内脏高动力,以期待更多关于交替RASS的研究。综上,针对PHT的治疗,我们要做到特异性,而不是简单的“一刀切”策略,期望针对肝硬化PHT发病和治疗有更深入的研究。
