基于细胞融合开发双基因共敲除技术
2021-03-18于鲁孟刘思思高晓冬藤田盛久
于鲁孟, 刘思思, 高晓冬, 藤田盛久
(江南大学 生物工程学院,江苏 无锡 214122)
近年来,针对哺乳动物细胞基因编辑的手段越来越多,包括设计锌指结构(ZFs)[1]、转录激活因子样效应物(TALEs)[2]、CRISPR/Cas9 技术[3-4],这些技术都可以特异性的敲除基因。 尤其是CRISPR/Cas9技术通过目标导向RNA 介导Cas9 蛋白造成DNA双链断裂[5],可以更精准的进行基因编辑,有效地缩短了基因编辑的周期和成本, 获得了广泛的运用。然而这些编辑手段都只能特异性编辑一个基因,目前有效编辑几个基因还有一定局限性。 已有相关文献报道[6-8],可以借助CRISPR/Cas9 技术和含多个目标导向RNA 的单个慢病毒质粒实现易转染细胞的多基因共敲除, 但是基因敲除效率取决于细胞类型。 因此仍需探索一种高效稳定的动物细胞多基因共敲除方法。
众所周知, 在酿酒酵母(Saccharomyces cerevisiae)的生命周期中,单倍体酵母细胞通过配对产生二倍体酵母细胞。 当营养缺乏时,二倍体酵母细胞形成孢子,从而再次产生酵母单倍体细胞[9]。在有丝分裂过程中,酵母细胞染色体发生重组并平均分配到两个子细胞中[10]。 若已敲除两个不同基因的两个单倍体酵母细胞经过配对和孢子形成,可以产生基因双敲除的酵母细胞[11]。 在哺乳动物细胞中相同或不同类型细胞可以融合并导致同核或异核形成,且亲本核保持离散并包含在由共享细胞质支持的单个细胞体中[12-14]。 不同种细胞间可以通过融合产生杂种细胞,获得多种目标表现型。 比如,将胚胎干细胞分别与神经干细胞、成纤维细胞或胸腺细胞融合的实验表明,通过细胞融合可以促进细胞向多能干细胞转化[15-16]。 通过细胞融合也可利用现有细胞直接获得新型细胞,而不必进行复杂漫长的基因编辑工作。 人类胚胎肾细胞 (Human Embryonic Kidney 293 cells,HEK 293),是一种特定的细胞系,最初来源于在组织培养中生长的人类胚胎肾细胞[17]。 HEK 293 细胞易于生长且表现出较高的转染效率。 HEK 293 细胞的基因组学、转录组学及蛋白质组学数据现已基本完整[18],因此以HEK 293 细胞为模型进行科学探索更具优势。
结合哺乳动物细胞基因编辑技术和细胞融合技术,以HEK 293 细胞株为模型建立了一种双基因共敲除技术。 为了便于鉴定细胞基因型,分别构建了 PIGA 敲除细胞株、PIGK 敲除细胞株、FUT8 敲除细胞株以及ST6GAL1 敲除细胞株。 PIGA 和PIGK均为糖基磷脂酰肌醇(glycosylphosphatidylinositol,GPI)生物合成的必须基因[19-20],可根据细胞表面GPI锚定蛋白表达情况鉴定基因敲除与否。 岩藻糖转移酶 8 是糖链结合核心岩藻糖的主要酶[21];α-2,6-唾液酸转移酶1 是糖链结合α-2,6 连接的唾液酸的必须酶[22-23],可根据细胞表面糖链结构判定基因敲除与否。由于现阶段没有以HEK 293 细胞为模型研究细胞融合条件的可供参考的数据, 故以PIGA 敲除细胞株和PIGK 敲除细胞株为模型优化细胞融合的条件。 依据优化后的细胞融合条件并结合CRISPR/Cas9 技术构建了 FUT8 和 ST6GAL1 双敲除细胞株。 本研究构建的动物细胞基因敲除技术可有效缩短构建双基因敲除细胞的时间。
1 材料与方法
1.1 细胞培养
大肠杆菌培养基:Amp 培养基: 胰蛋白胨,1.6 g/dL 酵母提取物质量分数1.5%,NaCl 质量分数0.5%。 灭菌后添加氨苄青霉素(Amp)使其终质量浓度为100 mg/mL。
SOB 培养基:胰蛋白胨2 g/dL,质量分数0.5%,酵母提取物质量分数0.5%,NaCl 质量分数0.5%,KCl 2 g/dL,MgCl210 mmol/L。
SOC 培养基:1 L SOB 中添加 10 mL 2 mol/L 葡萄糖溶液。
细胞培养基:
FBS 培 养 基 :500 mL DMEM 溶 液 (Gibco,Life technologies)中加入55 mL 胎牛血清。
Hyg 培养基: 向 550 mL 质量分数 10% FBS 培养基中加入2.2 mL 100 mg/mL 的潮霉素溶液。
BSD 培养基:向 550 mL 质量分数 10% FBS 培养基中加入0.55 mL 100 mg/mL 的杀稻瘟菌素溶液。
Puro 培养基:向 550 mL 质量分数 10% FBS 培养基中加入55 μL 100 mg/mL 的嘌呤霉素溶液。
人体胚胎肾细胞HEK 293 和HEK 293 T 细胞株培养条件均为37 ℃,CO2体积分数5%。
1.2 质粒构建
构建 pME-Hyg-EGFP 质粒。 以 pME18s-EGFP为模板,PCR 扩增绿色荧光蛋白基因片段(EGFP)。以限制性内切酶处理并纯化PCR 扩增片段,用连接酶(Ligation Mix,Takara)连接入 pME-Hyg 的 EcoRI和NotI 位点。
构 建 pX330 -EGFP -FUT8 -cr1 和 pX330 -EGFP- FUT8-cr2 质粒。 通过 E-CRISP 网站(http://www.e-crisp.org/E-CRISP/) 设计敲除 FUT8 的两对目标序列 FUT8-cr1 和 FUT8-cr2,qPCR 分别扩增这两对目标序列, 以限制性内切酶处理并纯化qPCR 扩增片段, 用连接酶连入 pX330-EGFP 的BbsI 位点。
构建 pX330-EGFP-ST6GAL1-cr1 和 pX330-EGFP- ST6GAL1-cr2 质粒。 同上。
构 建 Lenti-Cas9-Blast-FUT8-cr1 质 粒 。 以pX330-EGFP-FUT8-cr1 为模板,PCR 扩增 FUT8 目标序列 (FUT8-cr1)。 以限制性内切酶处理并纯化PCR 扩增片段, 用连接酶连接入Lenti-Cas9-Blast的NheI 位点。
构建 Lenti-Cas9-Puro 中间质粒。 以 Lenti-CRISPR v2-Puro 为模板,PCR 扩增嘌呤霉素基因片段(Puro)。 以限制性内切酶处理并纯化PCR 扩增片段,用连接酶(Quick cloning Mix)连接入Lenti-Cas9的 BamHI 和 EcoRI 位点。
构建 Lenti-Cas9-Puro-ST6GAL1-cr1 质粒。 以pX330-EGFP-ST6GAL1-cr1 为 模 板 ,PCR 扩 增ST6GAL1 目标序列(ST6GAL1-cr1)。 以限制性内切酶处理并纯化PCR 扩增片段, 用连接酶连接入Lenti-Cas9-Puro 的 NheI 位点。

表1 本文中所用引物Table 1 Primers of this stduy
1.3 细胞株构建方法
1.3.1 Lipofectamine 2000 介导HEK 293 细胞株稳定转染在6 孔板上接种HEK 293 野生型细胞,培养至细胞长到平板底面积的80%。 先将质粒酶切,然后按照Invitrogen 的说明书, 将线型质粒转染到HEK 293 野生型细胞。 待细胞长到满板,用含抗生素的培养基筛选,即可获得新型稳定细胞株。
1.3.2 Lipofectamine 2000 介导 HEK 293 细胞株瞬时转染在6 孔板上接种HEK 293 野生型细胞,培养至细胞长到平板底面积的80%。 按照Invitrogen的说明书, 将环型质粒转染到HEK 293 野生型细胞。 待细胞长到满板,用含抗生素的培养基筛选,即可获得新型稳定细胞株。
1.3.3 慢病毒介导HEK 293 细胞株稳定转染在6 孔板上接种HEK 293 T 细胞,培养至细胞长到平板底面积的80%。 将PMD-G,psPAX2 与重组质粒按照质量比 1∶1∶2 转染到 HEK 293 T 细胞, 将在HEK 293 T 细胞中产生慢病毒。 24 h 后,将含有病毒的培养基与质量分数10% FBS 培养基以体积比1∶1 混合,然后接入 HEK 293 野生型细胞。病毒转染24 h 后用含抗生素的培养基筛选1 周。 对细胞染色并用流式细胞分选仪S3e 分选未染色细胞,由此获得新型稳定细胞株。
1.4 细胞融合的实验操作
聚乙二醇是一种常用的细胞融合试剂。 日本血凝病毒外壳是日本凝血病毒去除遗传物质后获得的,也可以介导细胞融合。
以PEG 为诱导剂介导细胞融合。 收集细胞,按照杨建等[24]修改的PEG 的使用方法将 PIGA-KOHyg-GFP 细胞株和 PIGK-KO-BSD-BFP 细胞株(各5×106个细胞)融合,并用质量分数 10% FBS 培养基培养3 d, 通过流式细胞分析仪分析并确定细胞表型。
以HVJ-E 为诱导剂介导细胞融合。 收集细胞,按照GenomONE-CF EX 说明书中 HVJ-E 的使用方法将PIGA-KO-Hyg-GFP 细胞株和PIGK-KOBSD-BFP 细胞株(各 2×105个细胞)融合,并用质量分数 10% FBS 培养基培养3 d,通过流式细胞分析仪分析并确定细胞表型。
1.5 流式细胞分析
收集细胞,用磷酸盐缓冲液洗涤并重悬(细胞终浓度为 5×105个/mL)。CD59 抗体可识别 GPI 锚定蛋白CD59[25],因此PIGA 和PIGK 基因敲除的细胞用CD59 抗体及结合红色荧光蛋白的二抗染色。FUT8 敲除的细胞用结合荧光的凝集素LCA 或生物素凝集素LCA 染色[26],ST6GAL1 敲除的细胞用生物素凝集素 SSA(Bio-SSA,Cosmo Bio,Japan)染色[21]。采用流式细胞分析仪分析细胞的荧光信号。
1.6 荧光显微镜检测
用1 g/dL 的明胶溶液将垫片固定于24 孔板中, 然后加入大约3.3×104个细胞及质量分数10%FBS 培养基培养 1~2 d。 用 4 g/dL 的多聚甲醛固定液固定细胞然后加入氯化铵溶液。 将垫片置于一片干净的载玻片上并封闭,通过共聚焦荧光显微镜检测细胞的荧光信号。
1.7 核型分析
在6 孔板上接种待检测细胞并培养至细胞长至平板底面积的50%。 向平板中加入秋水仙胺(质量浓度1 μg/mL)培养3 h,使细胞的有丝分裂停止于染色体辨识性最高的分裂中期[27]。 用1 g/dL 的柠檬酸钠溶液处理以防止细胞黏连,用乙酸与甲醇的混合溶液(体积比为1∶3)固定细胞。 将细胞液滴于温热的载玻片后滴加Hochest 溶液使染色体着色,盖上盖玻片并用指甲油封闭。 通过共聚焦荧光显微镜观察细胞荧光信号。
1.8 细胞生长活性检验
收集并计数细胞, 将200 个细胞接种到96 孔板中用100 mL 质量分数10% FBS 培养基培养。4 h后加入 10 μL CCK-kit,1 h 后借助酶标仪 (450 nm波长)分析细胞活性。
2 结果与讨论
2.1 PIGA-KO-Hyg-GFP 细胞株和PIGK-KOBSD-BFP 细胞株的构建
通过稳定转染方法将抗潮霉素基因和绿色荧光蛋白(GFP)基因插入PIGA-KO 细胞株中,同时将抗杀稻瘟菌素基因和蓝色荧光蛋白(BFP)基因插入PIGK-KO 细胞株,从而便于用抗性培养基筛选细胞以及借助流式细胞仪和共聚焦荧光显微镜确定细胞类型。 流式分析结果显示PIGA-KO 细胞样本具有绿色荧光、无蓝色荧光且不能结合Anti-CD59 抗体,如图1(a)、(b),共聚焦显微镜结果显示细胞具有绿色荧光、无蓝色荧光,如图1(c);PIGK-KO 样本细胞具有蓝色荧光、 无绿色荧光且不能结合Anti-CD59 抗体,如图2(a)、(b),共聚焦显微镜结果显示细胞具有蓝色荧光、无绿色荧光,如图2(c)。从而确定PIGA-KO 细胞样本为PIGA-KO-Hyg-GFP 细胞株,PIGK-KO 细胞样本为 PIGK-KOBSD-BFP 细胞株。

图1 PIGA-KO-Hyg-GFP 细胞株的鉴定Fig.1 Identification of PIGA-KO-Hyg-GFP cell line
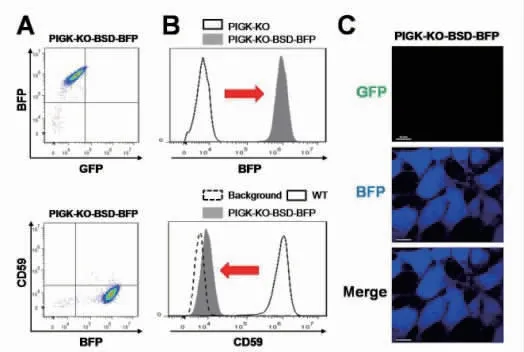
图2 PIGK-KO-BSD-BFP 细胞株的鉴定Fig.2 Identification of PIGK-KO-BSD-BFP cell line
2.2 PIGA-KO-Hyg-GFP & PIGK-KO-BSDBFP 融合细胞株构建
以 PIGA-KO-Hyg-GFP 细胞株和 PIGK-KOBSD-BFP 细胞株为模型构建融合细胞株。将上述两种细胞株混合并用PEG 处理。 流式分析结果显示,部分细胞株融合,如图3(a)。 限制性稀释含多种细胞株的混合样本,以期获得单克隆细胞株。 流式分析结果显示单克隆细胞株能够结合Anti-CD59 抗体,这表明已获得融合单克隆细胞株,如图3(b)。通过镜检可检测到绿色荧光信号和蓝色荧光信号,如图3(c),并且核型分析结果表明融合细胞中染色体数目近乎是PIGA-KO-Hyg-GFP 细胞株和PIGKKO-BSD-BFP 细胞株的染色体数目总和, 如图3(d), 这充分证明已获得 PIGA-KO-Hyg-GFP &PIGK-KO-BSD-BFP 融合细胞株且可以稳定培养。
2.3 细胞融合条件优化
依据现有融合方法获得HEK 293 融合细胞的效率极低,所以在此以HEK 293 细胞为研究模型进行细胞融合条件的优化,从而提高细胞融合效率。
2.3.1 以PEG 为融合试剂的细胞融合条件优化PEG 可以改变各种细胞膜结构,使两个细胞接触部分的脂质分子疏散并重组[2],从而引发细胞融合,如图4(a)。 由于 PEG 的相对分子质量不同,因而被分为不同的类型, 例如本实验中用到的Solar 4000、Merck 4000、Sigma 8000、Roche 1500 和 Solarbio 3350 5 种 PEG。首先以 PEG 为融合试剂,从 PEG 的种类,如图4(b);37 ℃培养时间,如图4(c);室温培养时间,如图4(d);PEG 浓度,如图4(e)共 4 个方面对细胞融合条件进行了优化。 由于每个实验中细胞的融合效率不完全相同,因此作者仅展示其中的代表性结果以说明其趋势。 得到最优条件为:混合两种细胞 (各 5×106个细胞) 后用预热的 PBS 和DMEM 清洗, 室温下缓慢加入1 mL 质量分数50%的PEG,室温培养5 min,加入预热的DMEM 溶液,37 ℃培养120 min,用预热的DMEM 溶液清洗细胞2 次,然后用质量分数 10% FBS 培养基培养 3 d。 经过条件优化,细胞融合效率达到了6%。
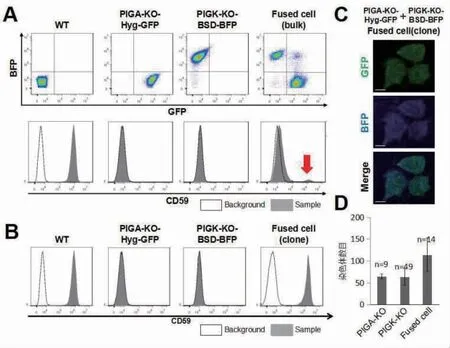
图3 PIGA-KO-Hyg-GFP 和PIGK-KO-BSD-BFP 融合细胞株的鉴定Fig.3 Identification of PIGA-KO-Hyg-GFP & PIGK-KO-BSD-BFP fused cell line

图4 PEG 介导的细胞融合条件优化Fig.4 Optimization of cell fusion by PEG
2.3.2 以HVJ-E 为融合试剂的细胞融合条件优化HVJ-E 可以像完整病毒一样使细胞膜发生改变从而引起细胞融合,如图5(a),却没有细胞致死性。也以HVJ-E 为融合试剂,从HVJ-E 使用剂量,如图5(b);是否需要离心,如图5(c);冰上培养时间,如图5(d);37 ℃培养时间,如图5(e)共 4 个方面对细胞融合条件进行了优化。 由于每个实验中细胞的融合效率不完全相同,因此作者仅展示其中的代表性结果以说明其趋势。 得到最优条件为:分别收集两种细胞(各2×105个细胞)后分别用PBS 清洗细胞一次并用25 μL 的融合缓冲液重悬细胞,将两种细胞溶液在2 mL 离心管中混合再加入2.5 μL 冰冻HVJE 溶液并用手轻敲离心管混匀, 冰上放置培养15 min,2 000 r/min 4 ℃离心 3 min,然后 37 ℃培养 25 min。 最后将细胞转移至6 孔板并加入预热的质量分数10% FBS 培养基培养3 d。经过条件优化,细胞融合效率达到了8.8%。
2.4 FUT8-KO-Hyg 细胞株和 ST6GAL1-KOBSD 细胞株的构建与融合
核心岩藻糖能被凝集素LCA 特异性识别并结合[26],因此可用结合荧光的凝集素(LCA-FITC)处理细胞并用流式细胞仪鉴定细胞表面糖链结构。 当唾液酸与半乳糖以α-2,6 连接时,唾液酸可被凝集素SSA 特异性识别并结合, 因此可用生物素凝集素(Bio-SSA)处理细胞并用流式细胞仪鉴定细胞表面糖链结构,如图6(a)。

图5 HVJ-E 介导的细胞融合条件优化Fig.5 Optimization of cell fusion by HVJ-E
2.4.1 FUT8-KO-Hyg 细胞株和ST6GAL1-KO-BSD细胞构建通过瞬时转染方法将pX330-FUT8-cr1和pX330-FUT8-cr2 质粒转入HEK 293 野生型细胞,并经限制性稀释获得FUT8-KO 单克隆细胞株。通过稳定转染方法将pME-Hyg 转入FUT8-KO 单克隆细胞株, 并用400 μg/mL Hyg 培养基筛选以期获得FUT8-KO-Hyg 细胞株。 流式分析结果表明细胞不能结合 LCA-FITC, 如图6 (b), 即获得了FUT8-KO-Hyg 细胞株。
通过瞬时转染方法将pX330-ST6GAL1-cr1 和pX330-ST6GAL1-cr2 质粒转入HEK293 野生型细胞,并经限制性稀释获得ST6GAL1-KO 单克隆细胞株。 通过稳定转染方法将 pME -BSD 转入ST6GAL1-KO 单克隆细胞株, 并用 10 μg/mL BSD培养基筛选以期获得ST6GAL1-KO-BSD 细胞株。流式分析结果表明细胞不能结合Bio-SSA, 如图6(c),即获得了 ST6GAL1-KO-BSD 细胞株。
2.4.2 FUT8-KO-Hyg 细胞株和ST6GAL1-KO-BSD细胞株融合以PEG 为融合试剂,依据细胞融合最优条件对FUT8-KO-Hyg 细胞株和ST6GAL1-KOBSD 细胞株进行融合处理。 用 400 μg/mL Hyg 和10 μg/mL BSD 培养基筛选后进行流式分析,结果显示样品中的绝大多数细胞均可结合LCA-FITC 和Bio-SSA,这表明绝大多数细胞已融合,如图6(d)。限制性稀释含多种表型细胞的混合样本,获得了单克隆细胞株。 通过流式分析单克隆细胞株,结果显示其均可结合 LCA-FITC 和 Bio-SSA,如图6(e),这初步表明获得了融合的单克隆细胞株。 通过电泳检测分析融合细胞株的FUT8 和ST6GAL1 两种基因型,结果显示融合细胞株中既有野生型的条带也有敲除型的条带,如图6(f)、(g)。 核型分析的结果表明样本细胞中染色体数目近乎是FUT8-KO-Hyg 细胞株和ST6GAL1-KO-BSD 细胞株的染色体数目总和,如图6(h),这充分证明已获得FUT8-KO-Hyg &ST6GAL1-KO-BSD 融合细胞株。
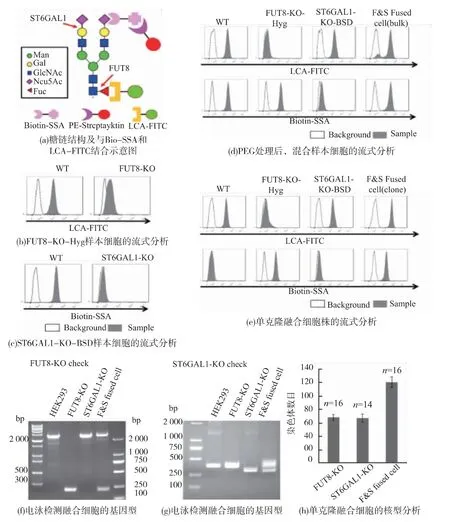
图6 FUT8-KO-Hyg 细胞株和ST6GAL1-KO-BSD 细胞株的鉴定及融合细胞的鉴定Fig.6 Identification of FUT8-KO-Hyg cell line,ST6GAL1-KO-BSDcell line and fused cell line
2.5 FUT8 和 ST6GAL1 的共敲除
质粒Lenti-Cas9-Blast 中有Cas9 基因,通过病毒转染可使HEK 293 细胞表达Cas9 蛋白, 在转入目的基因的目标序列的条件下可以实现目的基因的敲除[5],如图7(a)。 作者通过用生物素凝集素(Bio-LCA)和生物素凝集素(Bio-SSA)处理细胞并用流式细胞仪鉴定细胞表型,如图7(b)。 作者期望可以通过细胞融合获得基因双敲除细胞株。
2.5.1 FUT8-cr1-KO-BSD 细胞株和 ST6GAL1-cr1-KO-Puro 细胞株的构建、 鉴定及生长活性分析通过病毒转染的方法, 将 Cas9 和 BSD 基因及FUT8-cr1 单向引导 RNA 转入 HEK 293 野生型细胞株, 并用 10 μg/mL BSD 培养基筛选以期获得FUT8-cr1-KO-BSD 细胞株。 用流式细胞仪分析待测细胞,结果显示被检验细胞株不能被Bio-LCA 染色,如图7(c),说明FUT8 基因已被突变且不能表达,表明已获得FUT8-cr1-KO-BSD 细胞株。
通过病毒转染的方法, 将Cas9 和Puro 基因及ST6GAL1-cr1 单向引导RNA 转入HEK 293 野生型细胞株, 并用1 μg/mL Puro 培养基筛选以期获得ST6GAL1-cr1-KO-Puro 细胞株。用流式细胞仪分析待测细胞, 结果显示被检验细胞株不能被Bio-SSA染色,如图7(c),说明ST6GAL1 基因已被突变且不能表达,表明已获得ST6GAL1-cr1-KO-Puro 细胞株。
检测FUT8-cr1-KO-BSD 细胞株和ST6GAL1-cr1-KO-Puro 细胞株的生长活性并与HEK 293 野生型细胞进行比较,结果显示,3 种细胞的生长曲线基本一致,如图7(d)。 这表明 FUT8 或 ST6GAL1 的敲除不影响细胞的生长。
2.5.2 FUT8-ST6GAL1-DKO 细胞株的构建以HVJ-E 为融合剂, 依据细胞融合最优条件融合FUT8-cr1-KO-BSD 细胞株和 ST6GAL1-cr1-KOPuro 细胞株。 理论上两种细胞株融合以后,其胞内存在的目标序列产生的目标导向RNA 和Cas9 表达产生的蛋白会使对方细胞中的目的基因突变,如图8(a)。 借助流式细胞仪分析用 Bio-LCA 或 Bio-SSA 处理过的 FUT8-cr1-KO-BSD 细胞株和ST6GAL1-cr1-KO-Puro 融合细胞株, 结果显示:部分细胞未被Bio-LCA 染色,如图8(b),表明部分细胞中FUT8 被敲除; 绝大部分细胞未被Bio-SSA 染色,表明部分细胞中 ST6GAL1 被敲除,如图8(b)。通过计算,FUT8 的敲除效率为40.8%;ST6GAL1的敲除效率为99.9%。 而事实上,FUT8-cr1-KO-BSD& ST6GAL1-cr1-KO-Puro 细胞株既不能结合Bio-LCA 又不能结合 Bio-SSA,如图8(c),这说明融合细胞中的FUT8 和ST6GAL1基因均被突变且不能表达,即已构建FUT8-ST6GAL1-DKO 细胞株。

图7 FUT8-cr1-KO-BSD 细胞株和ST6GAL1-cr1-KO-Puro 细胞株的鉴定及生长活性检验Fig.7 Identification of FUT8-cr1-KO-BSD cell line and ST6GAL1-cr1-KO-Puro cell line and cell growth activity test
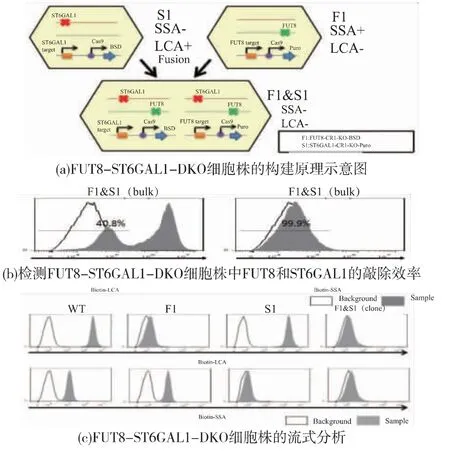
图8 FUT8-ST6GAL1-DKO 细胞株的鉴定Fig.8 Identification of FUT8-ST6GAL1-DKO cell line
3 结 语
作者构建了PIGA-KO-Hyg-GFP 细胞株和PIGK-KO-BSD-BFP 细胞株并将两者融合获得PIGA-KO-Hyg-GFP & PIGK-KO-BSD-BFP 融合细胞株。 通过对细胞融合实验条件的优化,使以PEG为融合试剂的细胞融合效率达到了6%,而以HVJE 为融合试剂的细胞融合效率达到了8.8%。 HVJ-E作为诱导剂进行细胞融合更具优势,如所需细胞数量少,不必大规模培养细胞,完成实验所需时间短,细胞融合效率高, 融合细胞死亡率低等,HVJ-E 作为诱导剂更有利于实验的进行。 构建了FUT8-KOHyg 细胞株和 ST6GAL1-KO-BSD 细胞株, 并以PEG 为融合试剂将其融合构建了FUT8-KO-Hyg 和ST6GAL1-KO-BSD 融合细胞株;构建了FUT8-cr1-KO-BSD 细胞株和ST6GAL1-cr1-KO-Puro 细胞株,并证明了FUT8 或ST6GAL1的敲除对细胞的生长活性没有影响。此外还以HVJ-E 为融合试剂将两者融合,并检测了基因敲除效率,最终构建了FUT8-ST6GAL1-DKO 细胞株。
与逐个敲除的常规方法相比,本研究可以直接通过细胞融合获得基因敲除细胞株,这有效地缩短了构建双基因敲除的哺乳动物细胞株所需的时间,也奠定了快速构建哺乳动物细胞多基因共敲除细胞株的基础。 但是融合细胞中存在染色体数目加倍且细胞生长相对缓慢的问题,后续研究将致力于此问题的解决。
