团簇NiMo3P电子性质的研究
2021-03-15王美玲方志刚井润田
王美玲, 方志刚, 秦 渝, 井润田, 廖 薇
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
随着社会经济的快速发展,人们对材料性能提出了更高的要求。过渡金属合金材料由于其优异的物理、化学性能受到了广泛的关注[1-2]。Ni-Mo系列合金应用十分广泛,其具有良好的耐腐蚀性、强耐磨性、较好的电化学性能及力学性能[3-4]。不仅如此,Ni-Mo合金还是目前应用最广泛的加氢脱硫催化剂之一[5],但随着现代工业技术的飞速发展,二元过渡金属合金的性能已经不能满足实验及工业应用的需求。为了进一步提高过渡金属合金的各项物理、化学性能,多元过渡金属合金已经成为科研界的研究热点[6-7]。文献[8]通过X-ray衍射仪(X-ray diffraction,XRD)及Jade软件对Ni-P及Ni-Mo-P镀层进行表征,通过浸泡腐蚀实验进行对比,发现在热处理温度相同时,Ni-Mo-P镀层的晶粒尺寸明显小于Ni-P镀层,且在高温退火时,Ni-Mo-P镀层的耐腐蚀性也远高于Ni-P镀层;文献[9]采用场发射扫描电子显微镜(field emission scanning electron microscope,FESEM)、能谱分析和XRD等方法研究了Ni-Mo-P/PCTFE涂层的表面形貌及化学组成,同时采用电位极化和电化学阻抗谱研究了涂层的耐腐蚀性,并利用原子力显微镜研究了合金表面的耐摩擦性能,测定了合金表面的拒水性,结果表明:Ni-Mo-P/PCTFE涂层的耐蚀性比普通合金有明显提高,且表面更加光滑,摩擦系数低,憎水性好。
因此本文基于Ni-Mo系列合金,引入非金属原子P进行掺杂,以文献[10]为基础,确定以团簇NiMo3P为模型进行多方面的研究,从而探究出团簇NiMo3P的微观电子性质。
1 模型和计算方法
根据拓扑学原理,运用密度泛函理论(density functional theory,DFT)[11],利用Gaussian09程序对二、四重态下团簇NiMo3P的20种初始构型进行优化计算,获得9种能稳定存在的优化构型,其中四重态4种,二重态5种[12]。利用Multiwfn程序提取各原子的电荷量,并利用Gaussian09程序提取各构型的电子自旋密度与各原子轨道的布居数等相关数据。在B3LYP泛函的条件下,采用Lanl2dz基组对Ni的最外层3d84s2价电子、Mo的最外层4d55s1价电子及P的最外层3s23p3价电子进行描述。P原子的核外电子排布为1s22s22p63s23p3,其价电子没有d轨道的存在,但大量实验表明,在计算过程中第三周期元素不仅存在d轨道,且其d轨道还作为价轨道参与s、p、d杂化成键[13],故P原子的布居数中可视为含有3d轨道的存在。
本文在B3LYP/Lanl2dz水平下,对Ni、Mo原子采用文献[14]提出的18-eECP双ξ基组(3s,3p,3d/2s,2p,2d);对P原子采用Dunning/Huzinaga双ξ基组(9s,5p/3s,2p),且P的极化函数[15]为0.55。以上所有计算均在启天计算机M4390上完成。
2 结果与讨论
2.1 团簇NiMo3P的构型与稳定性
以三角双锥型、四棱锥型和平面五边形构型为基础,改变不同原子的相对位置,设计出团簇NiMo3P的20种可能存在的构型,将20种构型进行优化并排除相同构型与含虚频的不稳定构型后,得到9种最终能稳定存在的优化构型,如图1所示,其中四重态4种,二重态5种。将能量最低的构型1(4)作为基准(设其能量为0),按能量由低到高将所有构型依次排序,各构型括号内的数字表示重态[16]。
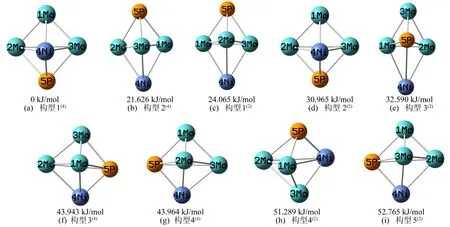
图1 团簇NiMo3P的优化构型图
从图1可以看出,团簇NiMo3P的9种优化构型皆为三角双锥型,这说明三角双锥型为团簇NiMo3P的优势构型。与四棱锥型和平面五边形构型相比,三角双锥型具有更好的稳定性,故团簇NiMo3P 9种优化构型的热力学稳定性均较为优异。其中构型1(4)和构型2(2)、构型2(4)和构型1(2)、构型4(4)和构型5(2)皆为不同重态下的相同构型。整体而言,在所有优化构型中,构型1(4)能量最低,热力学稳定性最好;构型5(2)能量最高,热力学稳定性最差。
2.2 团簇NiMo3P的电子性质
2.2.1 团簇NiMo3P各原子电荷量
根据团簇NiMo3P中各原子的电荷量可以有效判断团簇各原子间的电子流向。将各构型的电荷量相加可以发现各构型的电荷量总值为0,这说明团簇NiMo3P整体上呈电中性,电子从电荷量为正值的原子流出,流入到电荷量为负值的原子中,具体见表1所列。
由表1可知,团簇各构型的Mo原子电荷总量为正值,说明Mo原子为团簇NiMo3P的电子供体。除构型1(2)中的Ni原子电荷量为正值外,其余皆为负值,且构型1(2)的Ni原子电荷量值较小,故从整体上看,Ni原子为团簇NiMo3P的电子受体。在四重态构型中,P原子电荷量均为负值,表现为接受电子;而在二重态构型中情况较为复杂,构型2(2)、3(2)和5(2)P原子的电荷量为正,其余构型电荷量仍为负值,因此要分情况讨论电子流向。综上分析可知,对于构型1(2),其电子流动方向为Mo、Ni→P;在构型2(2)、3(2)和5(2)中,电子流向为Mo、P→Ni;其余构型皆为Mo→Ni、P。
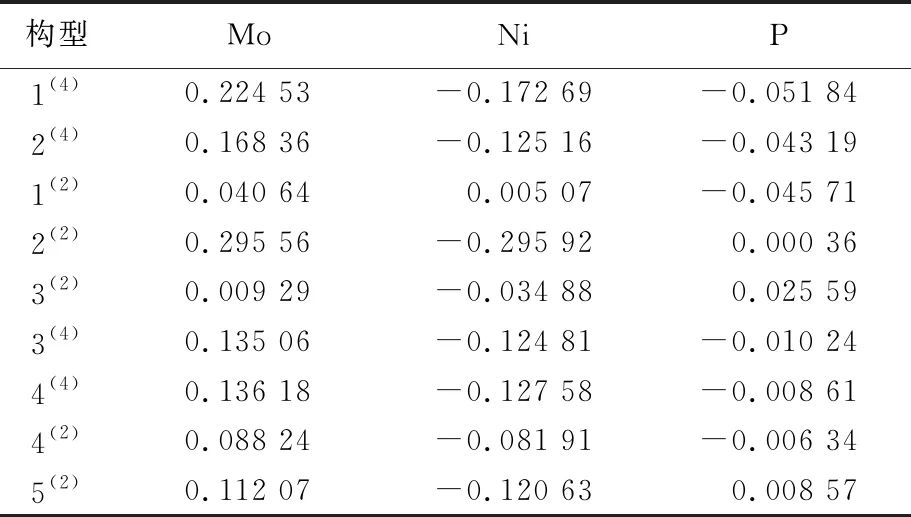
表1 团簇NiMo3P各原子电荷量
为了进一步观察各原子间电子流动性的强弱,作出各构型Ni、Mo、P原子的电荷总量变化趋势如图2所示。团簇各构型之间原子的电荷改变量是判断原子电子流动性强弱的重要依据,从图2可以看出,Mo、Ni原子的电子流动性大于P原子,故Mo、Ni原子为团簇NiMo3P内部电子流动的主要贡献者,且Mo、Ni原子的电荷量呈相反的变化趋势,因此团簇NiMo3P的电子主要是由Mo原子流向Ni原子。结合表1与图2,可以判断出团簇NiMo3P中各构型的电子流动性由强到弱依次为2(2)、1(4)、2(4)、4(4)、3(4)、5(2)、4(2)、1(2)、3(2)。其中不同重态下的同一构型1(4)和2(2)的电子流动性最强,但构型2(4)和1(2)、构型4(4)和5(2)间电子流动程度相差较大,说明团簇NiMo3P的空间结构能在一定程度上影响团簇内部的电子流动,但不起决定性作用。

图2 团簇NiMo3P中Ni、Mo、P原子的电荷总量
2.2.2 团簇NiMo3P各原子轨道布居数变化量
布居数表示原子在团簇中各轨道上的电子排布较原子本身电子排布的变化量。为了进一步研究团簇NiMo3P电子的微观流动状态,计算团簇NiMo3P各原子轨道的布居数变化量,结果见表2所列。布居数变化量为正值,表示该轨道有电子流入;变化量为负值,表示该轨道有电子流出。从表2可以看出,在团簇NiMo3P中Mo原子的5s轨道、Ni原子的4s轨道与P原子的3s轨道均为负值,表示有电子流出;除构型2(2)、4(4)和5(2)的Mo2原子4d轨道为负值外,其余构型Mo原子的4d、5p轨道、Ni原子的3d、4p轨道和P原子的3p、3d轨道均为正值,表示有电子流入。因此从整体来看,团簇NiMo3P内部电子主要是由各原子的s轨道流向各原子的p、d轨道。此外,从表2可以明显看出,在团簇NiMo3P中Mo原子的5s轨道、Ni原子的3d、4s、4p轨道和P原子的3s轨道布居数变化量较大,为团簇NiMo3P内部电子流动的主要贡献者。
从表2还可以看出,团簇NiMo3P中Mo原子的s、p、d轨道布居数总变化量均为负值,这说明在团簇NiMo3P中Mo原子为电子供体;Ni原子的布居数总变化量为正值,表示有电子流入,说明Ni原子为团簇NiMo3P的电子受体,这与2.2.1中所得结论一致。P原子情况较为复杂,在构型1(4)、2(2)、3(2)、3(4)中,P原子的布居数总变化量为负值,提供电子,在这些构型中电子从Mo、P原子流向Ni原子;在其余构型中,P原子的布居数总变化量为正值,电子从Mo原子流向Ni、P原子。P原子电子流动方向的不确定性恰恰说明团簇内部微观电子流向具有复杂性。

表2 团簇NiMo3P各原子轨道布居数变化量
2.3 团簇NiMo3P的电子自旋密度
2.3.1 团簇NiMo3P各原子的电子自旋密度
原子的电子自旋密度是影响团簇稳定性的重要因素之一,当原子的电子自旋密度分布明显时,可以判断团簇各优化构型的稳定性。团簇NiMo3P各原子自旋密度见表3所列,表3中,正值表示α电子出现的净概率密度;负值则表示β电子出现的净概率密度。从表3可以看出,构型1(4)~3(2)皆有一个Mo原子的电荷分布为β电子,2个Mo原子的电荷分布为α电子;构型3(4)~5(2)的Mo1、Mo3原子电荷分布为自旋向下的β电子,Mo2原子的电荷分布为自旋向上的α电子。最稳定构型1(4)的Mo2、Mo3电子自旋密度近似相同且较大,电子在这2个Mo原子间被离域平分,使得团簇NiMo3P的能量降低,对团簇稳定性起促进作用。对于能量相近的构型3(4)和4(4),其Ni原子与P原子的电荷分布均为自旋向上的α电子,且Ni、Mo、P 3种元素的电子自旋密度值分别近似,故构型3(4)与4(4)的稳定程度相近。
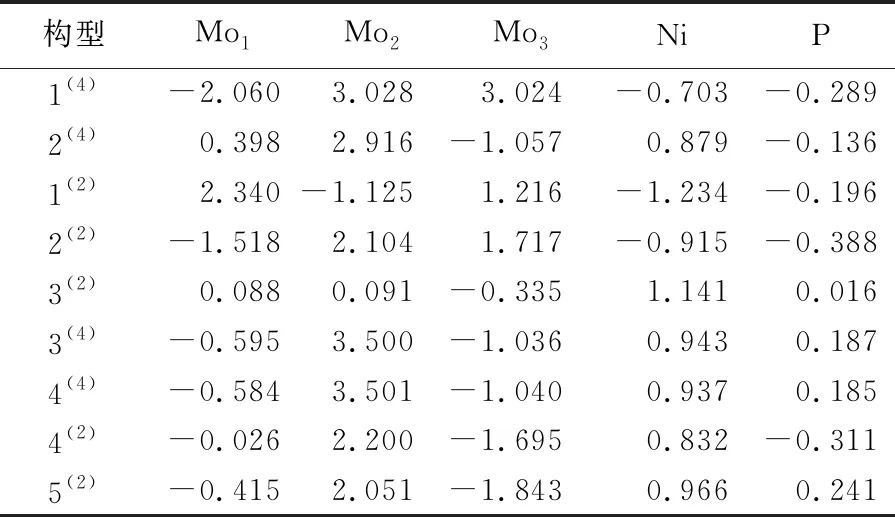
表3 团簇NiMo3P各原子的电子自旋密度
2.3.2 团簇NiMo3P各原子间电子自旋密度
团簇各原子间的电子自旋密度是影响成键强度的重要因素之一,原子间电子自旋密度的绝对值可以表征原子间的成键强度。成键强度越大、成键越均匀,团簇体系的能量越低,稳定性越好。团簇各原子间电子自旋密度见表4所列,其中正负分别表示成键时α、β电子的过剩情况,原子间电子自旋密度值为正,表示两原子间成键时α电子过剩;反之为负,表示两原子间成键时β电子过剩。
从表4中可以看出,最稳定构型1(4)的Mo1—Mo2和Mo1—Mo3、Mo2—Ni和Mo3—Ni、Mo2—P和Mo3—P间的电子自旋密度值分别相等,则构型1(4)的Mo1—Mo2和Mo1—Mo3、Mo2—Ni和Mo3—Ni、Mo2—P和Mo3—P成键强度相同,成键分布均匀,使得构型1(4)的能量降低,稳定性提高。其中Mo1—Mo2和Mo1—Mo3、Mo2—Ni和Mo3—Ni间均为α电子过剩,Mo2—P和Mo3—P间为β电子过剩。此外,构型3(4)和4(4)各原子间的电子自旋密度分别近似相同,成键强度相近,故而体系能量相近。且两构型中Mo1—Mo2、Mo2—Mo3、Mo2—Ni、P—Ni、Mo2—P间成键时均为β电子过剩,Mo1—Mo3、Mo1—Ni、Mo3—Ni、Mo1—P、Mo3—P间成键时均为α电子过剩。
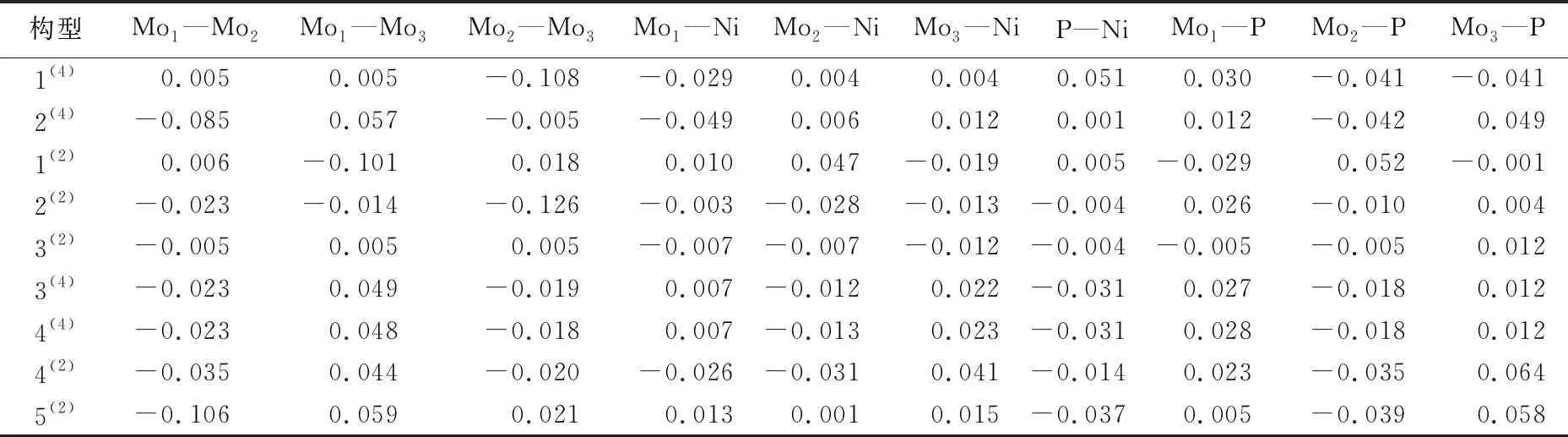
表4 团簇NiMo3P各原子间电子自旋密度
2.3.3 团簇NiMo3P稳定性规律分析
为了进一步探究团簇NiMo3P各优化构型的稳定性,作出团簇NiMo3P的电子自旋密度分布如图3所示,以此分析电子自旋密度分布对团簇稳定性的影响,并对其进行分类讨论。图3中,浅色表示α电子;深色表示β电子。
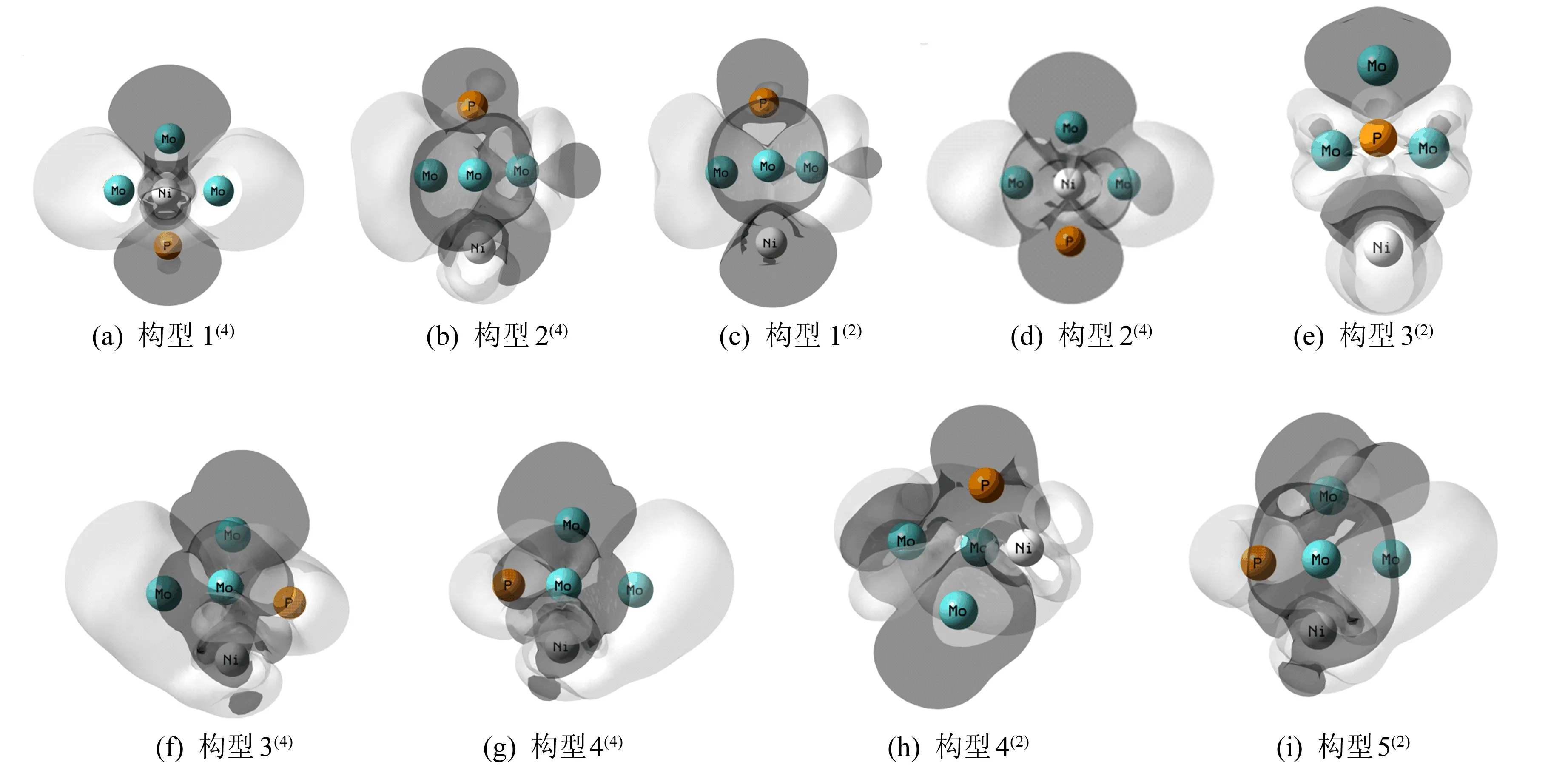
图3 团簇NiMo3P的电子自旋密度分布
第1类为不同重态下的同一构型1(4)和2(2)、构型2(4)和1(2)、构型4(4)和5(2)。
(1) 构型1(4)和2(2)。从图3可以看出,构型1(4)外围为α、β电子交替分布且高度对称,α、β电子的分布比例接近于1∶1,故构型1(4)的外围电子被离域平分,分布均匀,使体系能量降低,稳定性提高。构型2(2)的外围电子自旋密度分布较为对称,但对称程度不及构型1(4)高,且构型2(2)外围α、β电子的分布比例与构型1(4)相比差距较大。观察团簇内部α、β电子的重叠程度,构型1(4)内部的α、β电子重叠更为均匀。从表4可以看出,构型1(4)的Mo—Mo、Mo—Ni、Mo—P键间既有α电子过剩,又有β电子过剩;而构型2(2)的Mo—Mo、Mo—Ni键间仅有β电子过剩,故构型1(4)的内部电子重叠程度更好,稳定性更高。
(2) 构型2(4)和1(2)。构型1(2)与构型2(4)外围均为α、β电子交替分布。与构型2(4)相比,构型1(2)的外围电子分布更加均匀且对称,α、β电子的分布比例更加接近;构型内部电子重叠的均匀程度也是构型1(2)优于构型2(4),故从电子自旋密度分布的角度分析得到,构型1(2)的稳定性优于构型2(4)。但根据图1中各优化构型的能量可知,构型2(4)的稳定性优于构型1(2),与对电子自旋密度分布进行分析所得出的结论相悖,这是由于电子自旋密度分布仅仅是影响构型稳定性的因素之一,其在分析构型稳定性时可作为参考,但并不能起决定性作用。
(3) 构型4(4)和5(2)。构型4(4)与5(2)外围为α、β电子交替分布,其中构型4(4)的外围电子自旋密度分布更加均匀,且由表4可知构型4(4)的Mo—Ni原子间既有α电子过剩,又有β电子过剩;而构型5(2)的Mo—Ni原子间仅有α电子过剩,故构型4(4)的内部电子重叠程度更好,构型更加稳定。
第2类为其余构型3(2)、3(4)、4(2)。构型3(2)、3(4)、4(2)外部均为α、β电子交替分布。其中构型3(2)的外围电子分布高度对称,且从表4可以看出,构型3(2)内部的Mo1—Ni和Mo2—Ni、Mo1—P和Mo2—P间的电子自旋密度分别相等,Mo原子间的相互成键强度一致但重叠成键后剩余电子自旋方向不同,构型3(2)内部电子被离域平分,团簇稳定性较为优异,故在这3种构型中构型3(2)稳定性最好。比较构型3(4)和4(2)发现,构型3(4)的外围电子分布更加均匀,且构型3(4)的内部电子重叠程度更好,故构型3(4)的稳定性优于构型4(2)。
综上所述,团簇稳定性与团簇外围电子自旋密度分布的对称性、均匀程度及α、β电子的分布比例有关;团簇内部电子的重叠程度与均匀性亦是影响团簇稳定性的重要因素,但并不起决定性作用。其中构型1(4)外围电子自旋密度分布对称性、均匀性最好,α、β电子的分布比例最相近,内部电子重叠程度最好,故其稳定性最优。
3 结 论
本文从原子电荷量、布居数及电子自旋密度等角度分析团簇NiMo3P内部电子流动情况,以探究出团簇NiMo3P的电子性质及各优化构型的稳定性。
(1) 在团簇NiMo3P中,Mo、Ni原子的电子流动性远大于P原子,故Mo、Ni原子为团簇NiMo3P内部电子流动的主要贡献者,其中Mo原子为电子供体,Ni原子为电子受体,说明在团簇NiMo3P中,电子主要是由Mo原子流向Ni原子。
(2) 从原子轨道角度进行分析发现,在团簇NiMo3P的内部,电子是由各原子的s轨道流向p、d轨道,其中Mo原子的5s轨道、Ni原子的3d、4s、4p轨道与P原子的3s轨道布居数变化量较大,对团簇NiMo3P内部电子流动起主要贡献作用。
(3) 从电子自旋密度分布角度进行分析得知,构型外围电子自旋密度的分布比例、均匀性、对称性及构型内部电子的重叠程度均是影响构型稳定性的重要因素,但不起决定性作用。
综合多方面的分析确定,在团簇NiMo3P的所有优化构型中,构型1(4)的稳定性最好。本文从微观角度出发对团簇NiMo3P的电子性质进行了细致且深入的理论研究,并希望能够为进一步研究Ni-Mo-P体系提供有价值的参考。
