双环磺草酮的合成研究
2021-03-06杨辉斌
张 帆,梁 爽,孙 冰,李 斌,杨辉斌
(沈阳中化农药化工研发有限公司,新农药创制与开发国家重点实验室,沈阳 110021)
对羟苯基丙酮酸双氧化酶(HPPD)是植物质体醌和生育酚合成过程中的关键酶。抑制HPPD活性可导致质体醌和生育酚的正常合成途径被阻断,进而造成类胡萝卜素的生物合成减少、光合作用链电子传递受阻,最终导致植物中叶绿素被破坏,出现叶片白化症状,甚至死亡。因此,HPPD是理想的除草剂靶点。HPPD抑制剂类除草剂既可在苗前使用,也可在苗后使用,并具有高效、低毒、抗性低、环境友好以及使用安全等特点。自20世纪90年代以来,该类除草剂已成为农药化学研究领域的热点[1]。目前已开发的HPPD类除草剂主要有硝磺草酮、异唑草酮和苯唑草酮等[2-4]。2018年,在全球作物农药市场,除草剂的销售额为246.08亿美元,同比增长5.9%,占全球作物用农药市场的42.7%,其中HPPD抑制剂类除草剂增幅达11.1%,是除草剂中同比增速最快的产品类型[5]。
双环磺草酮属于双环辛烷类HPPD除草剂,其结构中有新颖的苯硫醚和双环结构,化学名称为3-(2-氯-4-甲基磺酰基苯甲酰基)-2-苯硫基双环[3.2.1]辛-2-烯-4-酮。2001年,双环磺草酮在日本登记并投放市场,商品名为Show-Ace,通过茎叶喷雾,用于水稻田和移栽水稻田一年生杂草的去除,有效剂量为168~252 g/hm2。双环磺草酮是内吸传导型除草剂,主要通过植物根茎部的吸收,导致新叶白化,故对萤蔺、异型莎草、扁秆藨草、鸭舌草、雨久花、陌上菜、泽泻、野慈姑、幼龄稗草、假稻、千金子等都具有较好的防效,尤其对萤蔺、鸭舌草防效突出。双环磺草酮持效期可达30~60 d,具有缓释效果,此外,其还具有处理适期宽和持效性长的特点[6-9]。
目前,关于双环磺草酮合成方法研究的文献报道较少,仅有关于葛发祥等[10]以降冰片烯为原料合成关键中间体双环[3.2.1]辛烷-2,4-二酮,进一步合成除草剂双环磺草酮原药的报道。笔者以2-氯-4-甲磺酰基苯甲酸为起始原料,依次经过酰氯化,酯化,重排,氯化,硫醚化5步反应合成双环磺草酮,并对酰氯化,酯化,重排3个关键步骤的工艺参数进行了系统研究,合成路线见图2。
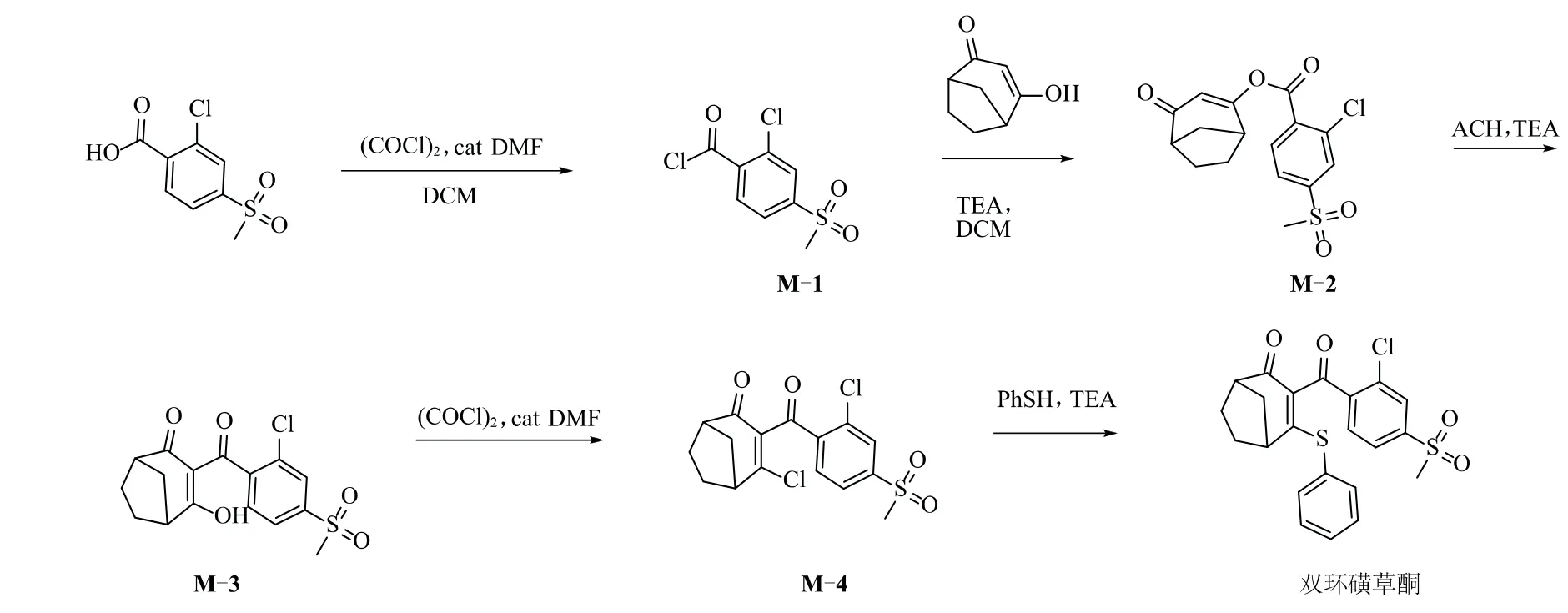
图2 双环磺草酮的合成
1 反应条件及合成研究
1.1 2-氯-4-甲磺酰基苯甲酰氯(M-1)的合成
文献中关于2-氯-4甲砜基苯甲酰氯的合成方法主要有2种。分别采用二氯亚砜或草酰氯作为酰氯化试剂[11-12],在不同的溶剂及温度条件下制备酰氯,得到的酰氯经浓缩后直接用于酯化反应。结果见表1。
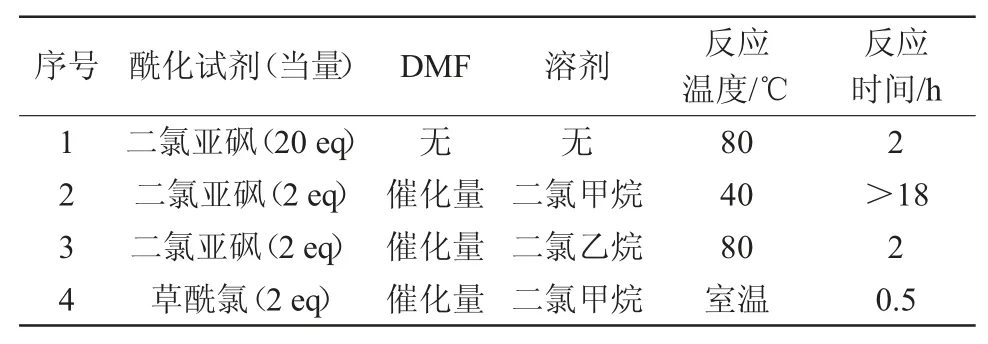
表1 2-氯-4-甲磺酰基苯甲酰氯反应条件
首先,尝试以二氯亚砜为溶剂的酰化条件(序号1),但原料2-氯-4-甲磺酰基苯甲酸在二氯亚砜中的溶解度较小,随着反应温度升高和反应的进行,原料逐渐溶解,反应时间约为2 h;其次尝试以二氯甲烷为溶剂,采用二氯亚砜为酰化剂,DMF为催化剂的体系进行反应。该体系反应进程十分缓慢,40℃过夜后仍有大量原料剩余(序号2);然后尝试以二氯乙烷代替二氯甲烷为溶剂,二氯亚砜-DMF体系进行反应的效果也不太理想(序号3);最后尝试以活性更高的草酰氯为酰化试剂,DMF为催化剂,二氯甲烷为溶剂的酰氯化体系。该体系具有极高的反应能力,在0.5 h左右,原料即可完全转化成酰氯,经减压脱溶后得到酰氯粗品,直接用于酯化反应。
1.2 中间体M-2的合成
该酯化反应采用经典的酰氯-二酮-三乙胺体系,以二氯甲烷为溶剂,考察了底物投料比对反应的影响,结果见表2。

表2 酯化条件
当苯甲酰氯过量时(序号1),以三乙胺做碱化,室温条件下反应4 h,双环[3.2.1]辛烷-2,4-二酮转化率可超过90%,但酰氯剩余较多,剩余的酰氯及苯甲酸需多次以饱和碳酸氢钠溶液洗涤方可去除,操作繁琐;将酰氯与二酮当量比变为1∶1时,即使过夜反应,2个原料仍有较多剩余(序号2);当二酮稍过量时,在相同的条件下,约3 h,酰氯即可实现较高的转化率,过量的二酮经10%NaOH溶液洗涤1次即可去除(序号3);进一步地提高二酮当量对酰氯转化率的提升无显著影响(序号4)。最终笔者采用酰氯∶二酮∶三乙胺当量比1∶1.1∶1.1的条件,以极高的转化率制备得到烯醇酯(M-2)。
1.3 中间体M-3的合成
文献报道的烯醇酯重排条件为三乙胺-丙酮-氰化氢体系[6]。该体系由于需要通入氰化氢气体,操作不便且危险性较大。三乙胺-丙酮氰醇体系是进行该重排反应常用方法。经实验发现,重排反应的主要副反应是烯醇酯分解为2-氯-4-甲磺酰基苯甲酸与双环[3.2.1]辛烷-2,4-二酮,结果见表3。
对该类反应的机制进行推测,如图3所示。
由于氰离子极强的亲核性,烯醇酯M-2很容易被氰离子进攻,形成高活性的苯酰腈中间体,若该中间体与体系中的水反应则生成苯甲酸副产物;若二酮以酮式进攻苯酰腈中间体则会形成稳定的重排产物M-3;若二酮的烯醇式进攻苯酰腈中间体则形成烯醇酯M-2,该M-2会继续被氰离子进攻,进入新一轮催化循环,直至形成稳定的苯甲酸或重排产物。由反应机理可知,重排与水解为1对竞争反应,虽然二酮的亲核性高于水,但在较高的温度下,反应选择性会降低。因此采用2种方案以避免副产物的形成:其一,降低体系中的水含量;其二,降低反应温度以提高选择性。最后采用无水硫酸镁干燥二氯甲烷作为溶剂,在10~15℃条件下滴加丙酮氰醇,再缓慢升温至室温,搅拌过夜,获得了较为满意的反应效果。
2 实验部分
2.1 仪器与试剂
主要试剂:所用试剂为市售化学纯或分析纯。
主要仪器:Mercury 300(Varian)核磁共振仪(TMS为内标),美国瓦里安有限公司;RY-1型熔点仪,天津分析仪器厂;Agilent 1100系列高效液相色谱,美国安捷伦公司。
2.2 关键中间体的合成步骤
2.2.1 2-氯-4-甲磺酰基苯甲酰氯(M-1)的合成
将2-氯-4-甲磺酰基苯甲酸1.30 g(5.55 mmol)、二氯甲烷30 mL加入到100 mL反应瓶中,加入1滴DMF,降温至0℃,缓慢加入草酰氯1.41 g(11.1 mmol),滴毕,室温搅拌0.5 h,减压蒸除溶剂及过量的草酰氯,得白色固体(M-1)1.3 g,直接用于下一步反应。
2.2.2 4-氧代双环[3.2.1]辛-2-烯-2-基2-氯-4-(甲磺酰基)苯甲酸酯(M-2)的合成
在250 mL反应瓶中加入双环[3.2.1]辛烷-2,4-二酮0.84 g(6.08 mmol)、三乙胺0.62 g(6.12 mmol)以及二氯甲烷40 mL降温至0℃,将上步制得的1.3 g M-1溶解在20 mL二氯甲烷中,缓慢滴加到反应瓶,加料完毕,室温下搅拌3 h,减压蒸除二氯甲烷,加入150 mL乙酸乙酯,以10%NaOH溶洗涤,有机相以饱和食盐水洗涤至中性,干燥,浓缩,得白色固体(M-2)1.85 g,直接用于下一步反应。
2.2.3 3-(2-氯-4-甲基磺酰基苯甲酰基)-4-羟基-双环[3.2.1]-2-辛烯-4-酮(M-3)的合成
往上步制得的1.85 g M-2中加入二氯甲烷40 mL、三乙胺0.85 g(8.40 mmol),降温至10℃,搅拌下加入2~3滴丙酮氰醇,体系在3 h内缓慢升温至室温,室温下搅拌15 h,反应液中加入1mol/L HCl溶液,用饱和食盐水洗涤、干燥、减压脱溶,残余物经柱色谱提纯(淋洗液为PE∶EA=1∶1)得到白色固体(M-3)1.12 g(3.16 mmol),三步收率为57%。1H NMR(300 MHz,CDCl3)δ:8.12-8.10(m,2H)、7.98-7.96(m,1H)、5.91(s,1H)、3.13(s,3H)、3.06-3.03(m,2H)、2.27(d,1H)、2.19-2.11(m,3H)、1.77-1.72(m,2H)。
2.2.4 3-(2-氯-4-甲基磺酰基苯甲酰基)-4-氯代双环[3.2.1]-2-辛烯-4-酮(M-4)的合成
在100 mL的单口瓶中加入1.12 g M-3(3.16 mmol)和二氯甲烷30 mL,搅拌下加入草酰氯0.62 g(4.88 mmol)和2滴DMF,加料完毕,室温搅拌4 h。减压蒸出有机溶剂,残余物加入50 mL乙酸乙酯,有机层依次用50 mL饱和碳酸氢钠溶液、50 mL饱和食盐水洗涤、无水硫酸镁干燥,减压脱溶,残余物经柱色谱提纯(淋洗液为PE∶EA=1∶1)得到白色固体(M-4)1.00 g,收率为89%。1H NMR(300 MHz,CDCl3)δ:7.96-7.78(m,3H)、3.06(s,3H)、2.35-1.72(m,8H)。
2.3 目标化合物双环磺草酮的合成
在100 mL反应瓶中加入0.80 g M-4(2.15 mmol)和二氯甲烷50 mL,依次加入苯硫酚0.26 g(2.36 mmol)和三乙胺0.25 g(2.47 mmol),室温搅拌4 h。反应液依次用50 mL饱和碳酸钠溶液、50 mL饱和食盐水洗涤、无水硫酸镁干燥,减压脱溶得黄色油液。向粗品中加入10 mL丙酮后,冷却至0℃,保温2 h,过滤,滤饼以丙酮洗涤,得到淡黄色固体(目标化合物)0.59 g,收率为61%。1H NMR(300 MHz,CDCl3)δ:7.92-7.49(m,8H)、3.09(s,3H)、2.18-2.06(m,2H)、1.87-1.78(m,4H)、1.59-1.56(m,2H)。13C NMR(75 MHz,CDCl3)δ:197.0、192.0、184.5、145.8、142.0、135.5、131.2、130.5、129.8、129.7、129.6、128.4、127.2、125.8、50.2、44.6、44.5、42.8、37.5、31.5、26.2。ESI-MS(m/z):447.0([M+H]+)、468.9([M+Na]+)。
3 结 论
笔者以2-氯-4-甲磺酰基苯甲酸为起始原料,经5步反应合成了双环磺草酮,对关键中间体的合成参数进行了优化,缩短了反应时间,简化操作,避免了副产物的生成。目标化合物的结构经1H NMR和MS确证。本研究对双环磺草酮类衍生物的结构改造具有重要意义。
