基于Linux平台构建单细胞转录组病毒信息分析系统*
2021-03-03陆庭希侯晨阳胡艳玲
陆庭希,侯晨阳,谢 兴,胡艳玲
(广西医科大学生命科学研究院,南宁 530021)
随着测序技术的飞速发展,分析处理测序数据信息已然成为现代生物研究的常规手段,然而传统分析流程过于碎片化,步骤繁琐且耗时较长。生物信息分析系统包括生物数据预处理和分析工具两大宏观部分[1]。通常所指的生物信息分析系统是包括收集并进行预处理相关生物数据(包括核酸序列、蛋白质序列、蛋白质结构或知识库),集成自主开发软件、脚本以及相关的公共开源的分析工具,可供用户进行数据处理、数据分析、生物信息挖掘等功能的统一分析平台[2]。针对当前数据量大、关联复杂、方法多样、工具软件繁多等突出的需求现状,构建整合的数据分析系统,集成已有的平台及特定软件集合,达到加速分析速度、减少分析过程中随机误差,降低分析流程复杂度的效果,显得尤为重要。现阶段常规分析方法仍以各个软件单独调参为主要手段,分析流程中包含的所有软件均需要使用者手工修改相关代码参数,保证其正确有效运行来得出期望的结果。局限性在于目前的分析方法代码复现率低,在不同的设备、不同的系统环境下、不同的数据集中难以复现,对于大规模、多样本数据分析,存在分析效率低、随机误差大、分析结果不稳定等问题。生物信息分析流程自动化、标准化是亟待解决的问题。因此,基于Linux 平台与单细胞转录组测序数据,整合现阶段分析流程所使用的相关分析软件,将其无缝串联起来形成一个标准化的快速分析流程,建立该病毒分析信息系统是非常必要的,以助于达到快速分析医学大数据的目的。本文以Linux 系统为基础,整合多个生物信息分析软件,辅以自动化脚本和相关数据库构建单细胞转录组病毒信息分析系统,现将结果报道如下。
1 单细胞转录组病毒信息分析系统构建过程
1.1 分析系统环境配置
本系统基于Linux(Version:3.10.0-693.el7.x86_64,builder@kbuilder.dev.centos.org)操作系统开发,硬件使用多台超级计算机(CPU:32 个Intel(R)Xeon(R)Silver 4110 CPU@2.10GHz)组成的分布式服务器/物理计算节点使用DDR4 2666MHz内存125G或250G/70TB 冗余硬盘阵列(RAID 1 方式),软件部分使用开源的Linux 系统Centos,并进行基本的系统设置及环境配置,分析软件的安装及环境配置。并且,由于科学计算任务对系统的稳定性和可靠性提出了较高的要求,所以分布式服务器安装在室温稳定且低于25oC 的机房环境下,保证其正常工作和运行[3]。环境配置如下:export PATH=$PATH:/public/software/sratoolkit/bin/;export PATH=$PATH:/$User_PATH/prepare_fastq_v1/;export PATH=$PATH:/public/software/samtools/bin/;source/public/software/ellranger-3.1.0/sourceme.bash
1.2 系统分层设计流程
单细胞转录组病毒信息分析系统采用分布式分层设计,其中包括上游分析层,下游分析层。每层内整合了相关生物信息分析软件及自动化脚本,见图1。其优势在于,各层相互独立,在分布式集群中每个模块可以在不同计算节点中处理任务,便于拓展以及防止过多任务投入导致的宕机。同时,这种分层设计不仅仅在于物理层面的划分,而包含逻辑层面的划分。不同层的模块可以在同一物理计算节点中运行,而同层模块中运行也可以由不同的物理计算节点来共同并行完成[4]。
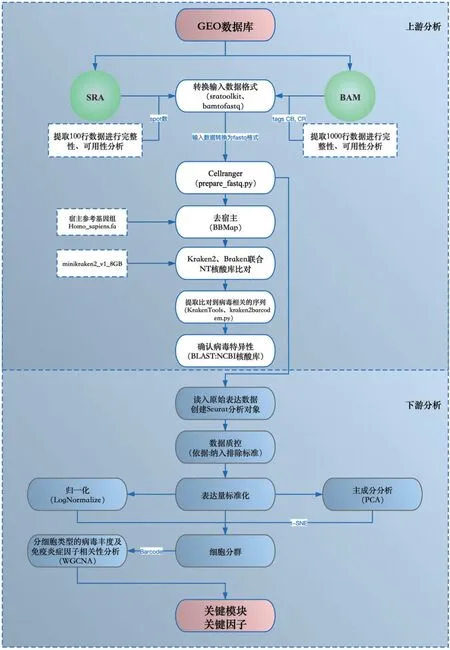
图1 单细胞转录组病毒信息分析系统分层流程图
1.2.1 上游分析层 结合系统分层流程图,对系统中的各模块进行介绍,各个模块集成软件,见表1。
数据源模块:系统数据来源,单细胞转录组测序数据来源可以是原始下机数据,同时支持GEO数据库(gene expression omnibus database)中的公共测序数据,数据通过SFTP 文件服务器进行上传及存储。该模块整合NCBI 中的sratoolkit:fastq-dump[5]和Bamtofastq-1.2.0,进行数据预处理。
数据处理模块:此模块应完成对源数据的上游分析处理,符合预处理要求的测序数据传入此模块。此模块主要集成了Cellranger、BBMap、Krak‐en2、Bracken、KrakenTools、BLAST[6]以及自主开发编写的Python脚本kraken2barcodem.py。
1.2.2 下游分析层 根据上游分析层获得的ma‐trix、features、barcodes 以及所提取对应物种的bar‐code序列,通过R语言的分析,可以确定比对所得的物种所在的细胞种类。该层集成了相关的R 包,来构建下游分析功能。R 包:Seurat(https://satijalab.org/seurat/)[7]、ggplot2、cowplot、Matrix、dplyr。
数据读入模块:此模块加载集成分析所需R包,并使用CreateSeuratObject 函数创建Seurat 对象,并提示用户对project/min.cell/min.featuresd 参数进行设置。数据读入支持10X 单细胞测序数据格式和原始表达矩阵格式的数据。
数据质控模块:数据质控是至关重要的环节,直接影响后续分析数据的可靠性。Seurat质控统计信息中包含每个细胞中的UMI数量和基因数,以及每个细胞中的线粒体基因占比;统计基因数、RNA、线粒体基因分布,并计算基因数与线粒体基因以及RNA 数量的分布相关性;筛选高质量细胞,尽量保证线粒体基因占比低于10%,表达的基因数至少大于200,测到的RNA 分子数在1 000~20 000 之间。以上质控结果以表图形式存储。
数据分析模块:此模块完成表达量的标准化、归一化与PCA、确定细胞类群分析PC、细胞分群、计算marker 基因的分析。表达量的标准化使用Log‐Normalize 算法,FindVariableFeatures 计算表达量变化显著的基因;归一化消除特定变量对整体数据的影响,如批次效应,线粒体数量的差异等。

表1 分析软件、R包的来源与功能
1.3 整合软件功能具体描述
上游分析部分:Sratoolkit由NCBI 提供,用于处理来自SRA 数据库中高通量测序数据的工具包,prefetch 模块可以批量下载数据库中数据集的原始数据;fastq-dump 模块可以将二代测序sra 格式的数据转换为分析所需的fastq 格式,其中双端测序文件使用--split-3 参数;Bamtofastq 是samtools 软件中的功能模块,其功能是从序列比对结果BAM 文件转换为fastq格式文件,对于双端测序数据需要使用-fq与-fq2 参数;Cellranger 是10X genomics 开发专门用于单细胞转录组测序分析的数据分析软件,可以完成数据拆分(cellranger mkfastq)、细胞定量(cell‐ranger count)、组合分析(cellranger aggr)、参数调整(cellranger reanalyze)等相关功能,将测序数据比对至参考基因组,完成细胞和基因表达定量并生成细胞-基因表达矩阵,基于此矩阵进行细胞聚类和差异表达分析;BBMap 是一种用于DNA 和RNA 测序reads 的比对工具,可以完成感知全局的拼接工作,它对基因组大小和contigs 数量没有限制,并且支持多线程来达到极快的索引速度;Kraken2 是一个基于k-mer 精确匹配算法的高精度序列分类软件,能够快速地将测序reads进行物种分类,进而注释出分析得到相关物种信息;Bracken(Bayesian Reestima‐tion of Abundance with KrakEN)是一种高精度统计方法,结合Kraken2 可以实现高准确度的测序数据物种分类分析,并计算物种丰度;KrakenTools 是一套适用于Kraken2/Bracken结果后续分析的脚本,在系统中整合了从kraken 结果中提取指定序列的功能;BLAST(Basic Local Alignment Search Tool)是在蛋白质数据库或DNA 数据库中进行相似性比较的分析工具,BLAST能迅速与公开数据库进行相似性序列比较,从而得出Coverage 值和Identical 值,BLAST结果中的得分是对相似性的统计说明,可以完成指定序列的特异性检验。
下游分析部分:Seurat 是一个R 包,其功能为识别和解释单细胞转录组数据中的异质性来源、整合了不同类型的单细胞数据的函数、单细胞转录组数据中的细胞质控、聚类、差异分析、细胞注释、以及结果可视化;ggplot2 是用于绘图的R 拓展包,整合在系统中为下游分析结果提供数据可视化;cowplot是一个绘图插件,可以为ggplot2提供出版级别的主题,优化可视化结果;dplyr 是一个用于数据清洗和处理的R包,整合到系统中,为输入数据的分析处理提供支持。
2 系统功能实现及测试结果分析
2.1 数据收集与预处理
当前信息分析系统整合了数据收集与预处理所需的软件,部署于服务器中,对软件完成封装。将sra、bam 格式测序数据转换为fastq 格式数据,并使用prepare_fastq.py 提取sra 格式数据的前100 行、bam 格式数据的前1000 行进行测试,判断其可用性,当数据不可用时,弹出提示并中断程序。对于sra 格式数据:如果spot 数为1,则判断该数据不可用;如果spot数为2,则spot1为read1,spot2为read2;如果spot 数 为3,则spot1 为index,spot2 为read1,spot3 为read2。同时检查read1、read2 长度,如果长度低于16,则不是barcode,判断该数据不可用;read2 长度要求不低于50 bp。对于bam 格式数据:检查tags CB,CR 是否完整,如果CR,CB 低于一定比例,则判断该数据不可用。符合预处理标准的数据,方能传入下一模块进行分析。
2.2 分析系统功能分析
2.2.1 参考库下载与用户数据管理 在构建此分析系统时,需要高效整合数据分析所用的参考库与参考基因组,以达到系统高效分析的效果。使用Cellranger 所需的人类参考基因组GRCh38,从Cell‐ranger 官 网(https://cf.10xgenomics.com/supp/cellexp/refdata-gex-GRCh38-2020-A.tar.gz)上获取已构建好的索引文件;BBMap 所使用的参考基因组为Homo_sapiens.GRCh38.dna.prmary_assembly.fa.gz(ftp://ftp.ensembl.org/pub/release-101/fasta/homo_sa‐piens/dna/);Kraken2 所使用的参考库为minikrak‐en2_v1_8 GB(http://ccb.jhu.edu/software/kraken2/in‐dex.shtml?t=downloads)。用户可依据不同任务自建工作文件夹在系统目录中,分析结果默认保存在系统目录中,若用户自建目录,则存放于用户指定目录下。
2.2.2 系统数据分析 在构建的单细胞转录组病毒分析系统中主要完成两个主要任务:一是从GEO数据库中获取单细胞测序数据,转换格式为fastq,完成质控等数据预处理,并与参考基因组比对去除宿主,在此基础上,联合Kraken2 与Bracken 对测序reads 进行物种注释,寻找病毒reads,并提取指定病毒序列和barcode序列,进行Blast验证其特异性,完成上游分析;二是对所获得的matrix、features、bar‐codes 以及所提取的对应物种的barcode 序列,通过R 语言分析来确定比对的物种所在的细胞种类,对细胞进行分群并计算marker 基因。其中,图2 展示了标准化之后的整体表达水平。图3展示了PCA分析从大量的基因表达信息中,提取了对整体基因表达量影响最大的效应,命名为PC1,PC2,基因之间的表达差异表现在PC1、PC2数值上的差异。图4以2D 点图展示了PCA 的结果,其中每个点代表了每个细胞。图5 用热图展示了PCA 的结果。图6 为聚类分析后的TSNE 的分群展示,其中每个颜色代表了一个cluster 后鉴定到的一种细胞群,散点代表了每个细胞,图中数字则代表了该群的cluster编号,由图中可知共分为了8 个不同的细胞群。图7 展示了子群中的marker 基因的表达水平,能更为直观地看到marker 基因在细胞中的表达水平,其中颜色越深则表示该基因在这些子群或细胞中的表达越高。图8使用小提琴图展示这几个marker基因在子群中的相对表达水平,可以衡量该基因作为子群marker基因的特异性。图9以热图的形式展示了marker基因在所有子群中的表达水平,并按照cluster 编号进行排序,图中的颜色表示了表达水平,黄色表示高表达。

图2 表达量标准化

图3 PCA结果-1

图4 PCA结果-2

图5 PCA结果-3

图6 TSNE细胞分群

图8 单个标记基因子群表达水平小提琴图
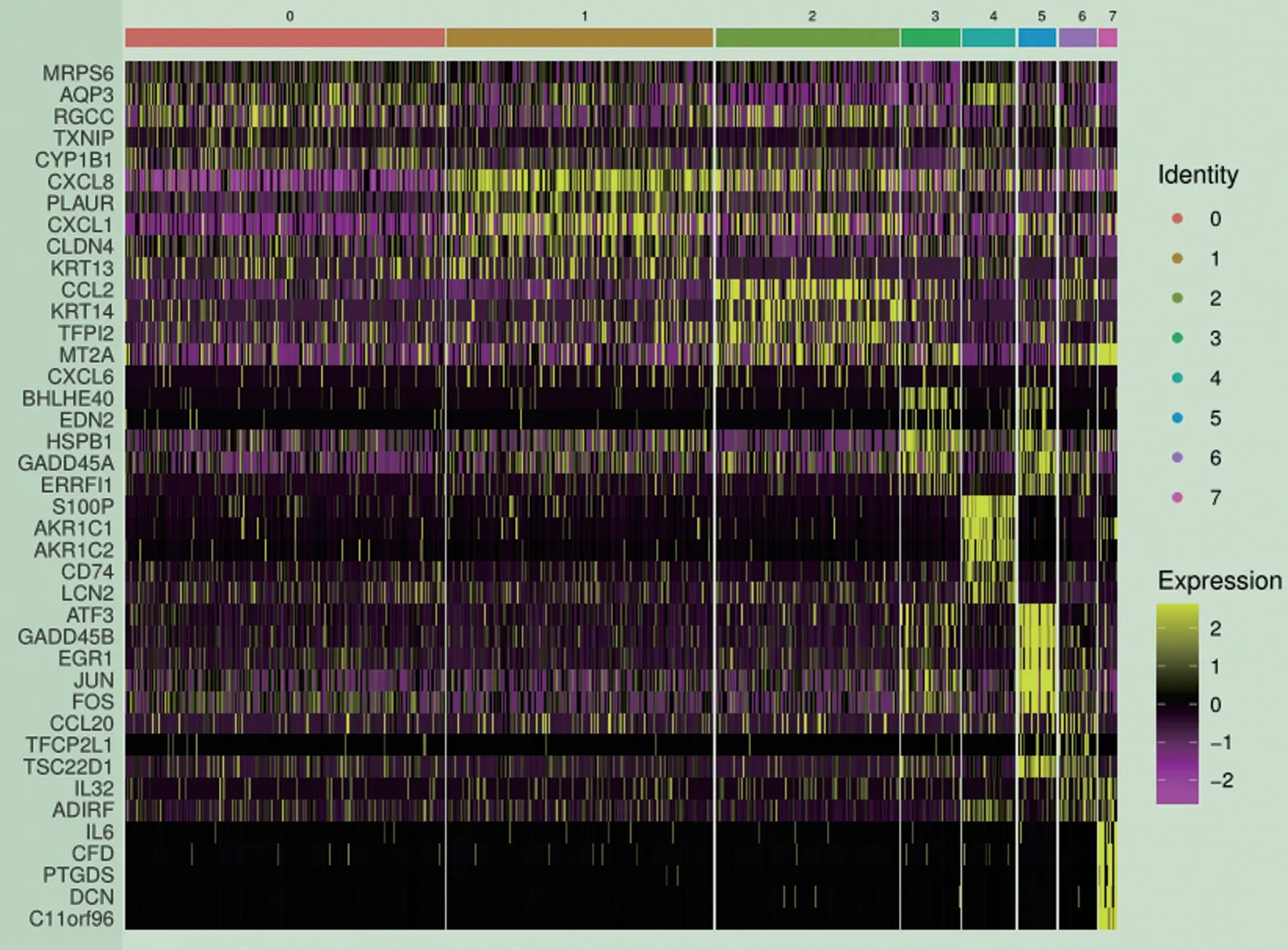
图9 Top marker基因热图
3 讨论
本文整合了多个可免费获得的生物信息学分析软件,辅以自主开发的自动化脚本,在Linux 平台上构建了可完成大数据、多任务单细胞转录组病毒信息分析的系统。应用该系统,我们已成功分析并获取到多组测序数据中病毒信息,得到了许多有意义的结果。
通过对本次构建的单细胞转录组病毒信息分析系统的深入研发,在不同生信分析软件之间建立了接口和构建了流程,给用户提供了完整、易用的分析平台,帮助用户跳过繁琐的软件安装及环境配置,简化分析流程,提高了数据处理的效率和处理的可行性[8]。传统的生物信息分析模式是单任务处理模式,对于大量测序数据来说,碎片化分析工作过于繁琐,构建本系统来分析则大大减轻了复杂程度。在大数据时代,高效分析显得尤为重要。本系统处理任务时,只需要将完整的单细胞转录组测序数据上传至指定工作目录,运行Python 自动化脚本即可依照预设流程,完成从数据读取质控到分析计算结束的工作,并通过SFTP 文件服务器下载所需的结果文件。应用本系统可以降低分析人员使用Linux 环境的难度,也避免了多用户在服务器直接操作带来的宕机风险和不安全因素,可以最大化服务器的工作效率和稳定性[9]。
本研究开发的单细胞转录组病毒信息分析系统仍存在一定的局限性。从理论方法层面上看,测试所使用的数据集数量比较少,还有待增加。系统最大负荷运算能力尚未评估,系统满负荷稳定性尚未评估,此方面后续研究需改进[10];使用层面上,需要使用者具备一定的Linux 使用基础以及生物信息学知识,个别参数针对不同类型来源数据需调整,还未开发人性化和谐美观的使用交互界面,系统美观性还存在不足。
猜你喜欢
杂志排行

广西医科大学学报的其它文章
- Effect of miR-200a-3p targeting transthyretin(TTR)on the biological behavior of placental trophoblasts in hypertensive disorder complicating pregnancy
- 沙利度胺对重型β地中海贫血患者红系细胞γ珠蛋白基因表达及分化的作用研究*
- CEMIP调控Src/AKT/STAT3信号通路对非小细胞肺癌上皮间质转化及细胞侵袭、迁移的影响*
- 乳铁蛋白对正畸复发移动过程中牙周改建的影响*
- 秦皮苷对脂多糖诱导的软骨细胞氧化应激和炎症因子的影响*
- miR-133b-3p对大鼠脑缺血/再灌注损伤的影响及其作用机制*