4类碳氢燃料的高温燃烧反应简化建模
2021-02-22梁兴雨刘志伟王昆朱仕皓王晓慧沈位
梁兴雨,刘志伟,王昆,朱仕皓,王晓慧,沈位
(天津大学内燃机燃烧学国家重点实验室,300072,天津)
大分子液态碳氢燃料由于体积能量密度和质量能量密度的平衡以及良好的存储运输特性,在可预见的未来依然是航空航天和地面交通运输等领域推动现代社会发展的主要能源。碳氢燃料的主要供能方式是燃烧,因此建立能够准确描述其燃烧特性的动力学模型对进一步提高发动机燃烧效率和降低污染物排放至关重要。传统汽油、柴油、航空煤油及多数可再生燃料等发动机实际使用的燃料通常是含多种大分子碳氢化合物的混合物;基于分子结构中化学官能团的不同,碳氢燃料可分为直链烷烃、支链烷烃、环烷烃和芳香烃等。与其他化学反应类似,燃烧反应非常复杂。基于详细化学反应机理建模的大分子碳氢燃料的燃烧反应动力学模型通常极为复杂,可包含成百上千个化学组分和成千上万个基元反应。例如,汽油典型化合物正庚烷和异辛烷、航空煤油组分正十二烷、柴油组分正十六烷,上述碳氢燃料的详细模型所包含的化学组分均达600~2 000个、基元反应达3 000~10 000个[1]。显然,准确的实验测量和可信赖的理论计算很难满足详细反应模型所涉及的大量热力学、动力学和输运参数,其建立过程很大程度上依靠预估、类比等经验或半经验方式。
同时,规模庞大、详细的燃烧反应模型只有大幅简化,方能耦合计算流体力学(CFD)以用于对真实发动机系统的仿真模拟,因而当前反应动力学模型的简化方法如关键成分分析法、反应路径分析法、敏感性分析法、误差传播直接关系图谱法、计算奇异摄动法、近稳态分析法等得到了长足发展[2-3]。为确保简化模型的保真性和准确性,以上方法常联合使用。尽管如此,可有效耦合CFD的燃烧反应模型依然需要进一步简化,以减缓或消除数值计算中化学反应与流体耦合带来的强刚性问题,并有效降低计算资源和费用。根据对燃烧反应物理过程的深入分析,在建模初始对其简化处理,再结合当前较为成熟的模型简化方法,是实现燃烧反应模型进一步简化的最有效方式之一。
发动机或燃气轮机内的燃烧通常是高温(1 000 K或以上)化学反应过程。在该反应条件下,大分子碳氢燃料的燃烧过程在时间和空间尺度均存在双区特性[4-5]。具体来讲,母燃料首先快速热解转化为小分子中间产物,之后中间产物发生较慢的氧化反应。基元反应特征时间分析可较好地解释以上高温燃烧过程的物理特性。同时,结合近稳态分析方法而发展的简化建模方法可显著降低大分子碳氢燃料转化为小分子中间产物的基元反应数量的规模。当前对小分子中间产物(又称基础燃料,通常包括氢气(H2)、C1~C4化合物以及苯(C6H6)和甲苯(A1CH3)等单环芳香烃)燃烧化学反应的基础研究主要基于较为完备的实验和理论研究方法,其动力学模型也较为成熟。
基于以上分析的两步反应过程的简化建模方法已经用于描述辛烷异构物的热解和氧化特性[4]。本文将该简化建模方法拓展至对较为复杂的环烷烃和芳香烃高温燃烧化学的描述,以进一步验证该方法的适用性。具体来讲,以正辛烷、异辛烷、甲基环己烷和正丁基苯为研究对象,它们代表真实燃料中4类官能团的大分子碳氢化合物;采用上述简化建模方法对这4类碳氢燃料建立简化反应动力学模型,并以热解反应和氧化反应的中间产物分布、点火延时时间和层流燃烧速率等基础燃烧实验数据对其进行验证。
1 模型构建
1.1 简化建模方法概述
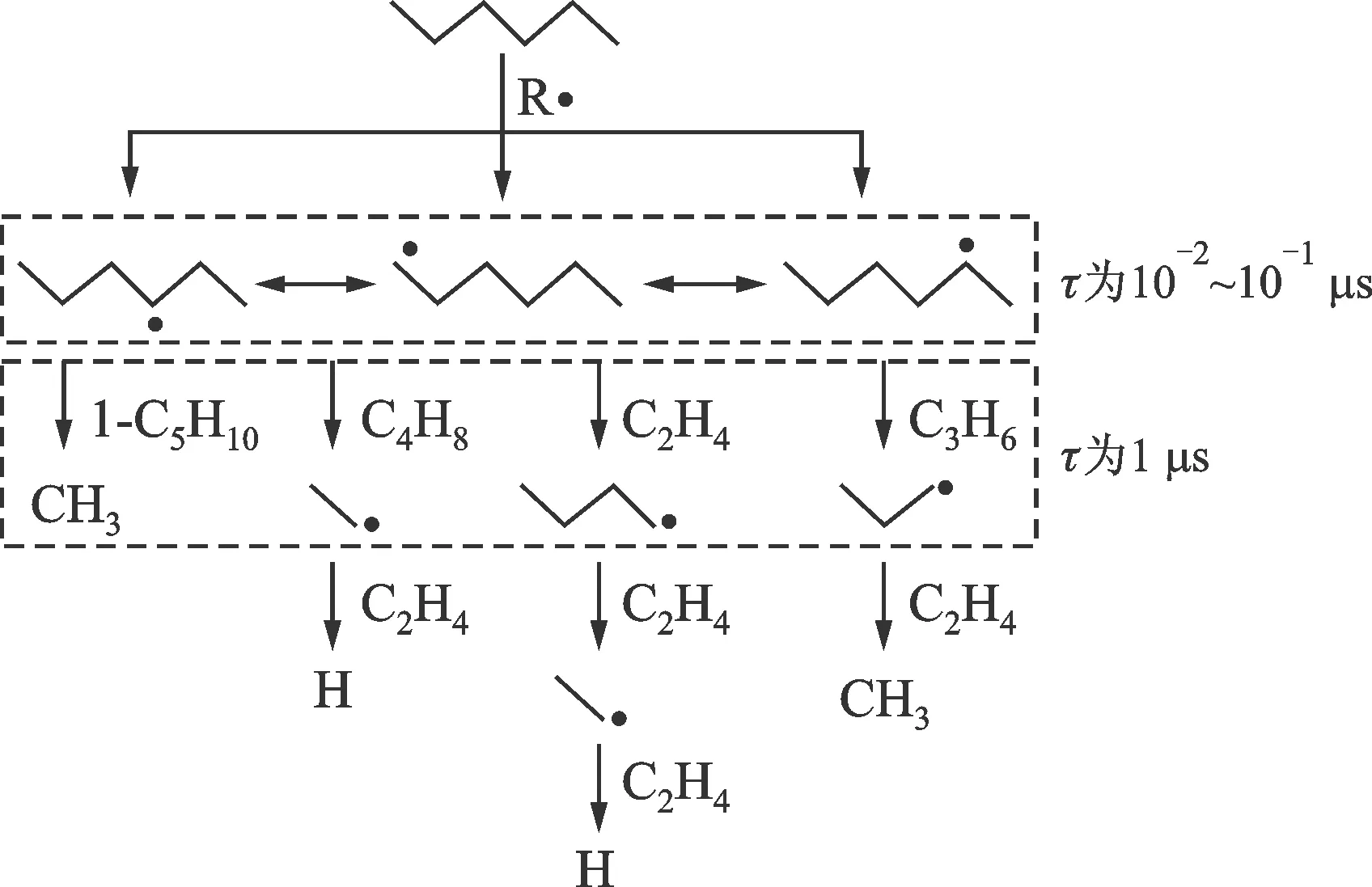
图1 高温热解反应过程和特征时间尺度
高温条件下,大分子碳氢燃料的燃烧过程分为先后进行的两个部分,即母燃料分子本身的快速热解反应和热解产物较慢的氧化反应。基于该燃烧物理过程,采用反应特征时间分析和近稳态分析的方法,可对该物理反应过程做简化建模[4]。如图1所示,以直键烷烃正己烷(n-C6H12)为例,由脱氢反应产生的3种烷基自由基,通过氢转移反应进行异构化反应进而达到热力学平衡。基于五环或六环过渡态结构的氢转移反应具有较低的能垒(分别为92和63 kJ/mol),其反应特征时间为0.01~0.1 μs。烷基自由基的C—C键β位断裂产生较小的烷基自由基(甲基或氢原子通过脱氢反应生成甲烷和氢气)或轻烯烃(包括乙烯、丙烯、丁烯等),但具有较高的能垒(146 kJ/mol),因而反应特征时间也较长,尺度为1 μs。以上分析符合准稳态假设,即正己烷脱氢反应后,其烷基自由基C—C键β位断裂是其裂解的决速步;平衡浓度的自由基决定了小分子烯烃产物的分布。基于上述基元反应特征时间分析和准稳态假设,采用7个总包反应(1个燃料C—C键离解反应和6个脱氢反应)描述母燃料通过热解/氧化热解反应迅速转化为小分子中间产物的反应过程(即热解反应简化模型)。小分子中间产物进入火焰燃烧区进行剧烈的氧化还原反应,实际控制整个燃烧过程的放热和终产物水和二氧化碳的生成。该过程较慢,需用详细的基础燃料氧化反应模型描述。
以上方法已经用于直链和支链烷烃的高温燃烧化学反应的描述[3]。本文将该方法扩展至化学反应动力学更复杂的环烷烃(以甲基环己烷为例)和芳香烃(正丁基苯)的高温氧化反应,进而探究不同官能团碳氢化合物对各自宏观燃烧特性的动力学控制作用。
1.2 动力学、热力学数据和输运参数的确定
7个总包反应的动力学参数来自各单步反应[6-7],涉及的化学组分热力学数据使用官能团贡献法获得[8]。表1~3所示为上述4类燃料热解反应简化动力学模型,其中R代表H·、CH3·、OH·、HO2·、O·和O2。本文选择USC-Mech-Ⅱ模型[9]作为基础燃料详细模型,直接用于对正辛烷、异辛烷和甲基环己烷的描述。对正丁基苯,其氧化和热解反应的中间产物实验数据显示[10-11],反应过程中生成了大量乙基苯(A1C2H5)、苯乙烯(A1C2H3)等芳香烃中间产物,而USC-Mech-Ⅱ模型对该类物质及其化学反应的描述不完善,因此,本文加入了部分化学反应,用以描述该类芳香烃物质的消耗。化学反应包括新产生自由基A1CH2·、A1C2H4·参与的正丁基苯脱氢反应及上述R、A1CH2·和A1C2H4·自由基参与的乙苯(A1C2H5)的脱氢反应。需要指出的是,补充该部分反应的目的是阐述该简化建模方法对烷基芳香烃氧化反应的可行性,未来可进一步完善其动力学描述。

表1 正辛烷和异辛烷高温热解反应简化动力学模型

表2 甲基环己烷高温热解反应简化动力学模型
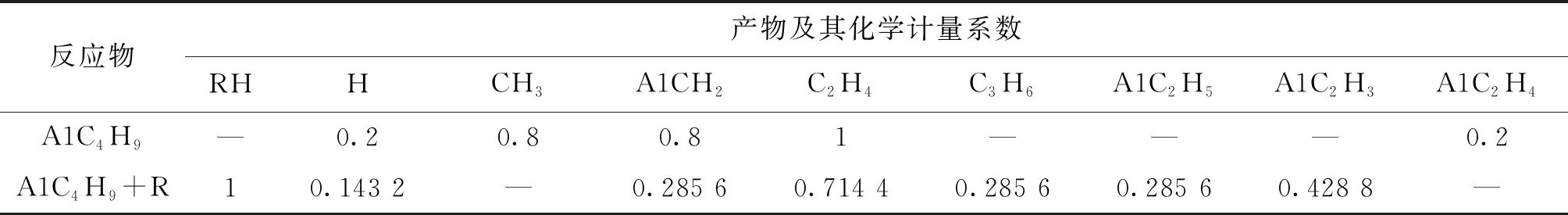
表3 正丁基苯高温热解反应简化动力学模型
由于表1~表3所示总包反应为不可逆反应,因此母燃料的热力学数据不会对动力学产生影响,但其会影响燃烧系统绝热温度等。母燃料的热力学数据和输运参数来自劳伦斯实验室详细机理(LLNL)或JetSurF v1.1详细机理,或通过官能团贡献法确定。
1.3 动力学模型的计算
4类燃料(即正辛烷、异辛烷、甲基环己烷和正丁基苯)的简化动力学模型在温度T=800~1 400 K、压力p=0.01~5 MPa、当量比φ=0.5~∞范围内均适用。所有动力学模拟均在动力学软件Ansys@ChemKin Pro中进行,采用的模块包括活塞流反应器(PFR)、射流反应器(JSR)、零维全混流反应器和预混层流燃烧火焰。使用来源较宽广实验条件的基础燃烧实验数据,包括高温热解和氧化反应中间产物分布、点火延时时间和层流燃烧速率等,对简化模型进行验证。作为参照,本文同时选取LLNL详细机理[12-14]进行对比验证。
2 结果与讨论
2.1 热解反应和氧化反应的重要中间产物
2.1.1 正辛烷 图2和图3分别对比了正辛烷(n-C8H18)氧化反应和热解反应主要中间产物的实验值与模拟值。其中,氧化实验数据来自流动管反应器系统[15],其具体条件为φ=0.99,T=1 076 K,p=0.1 MPa,nN2∶nO2∶n燃料=66∶1.2∶0.095。实验及模拟数据包括燃料转化及主要中间产物,后者包括乙烯(C2H4)、丙烯(C3H6)、甲烷(CH4)、1-丁烯(1-C4H8)和氧化终产物,即一氧化碳(CO)和二氧化碳(CO2)。热解实验数据来自射流搅拌反应器系统[16],其具体条件为停留时间τ=0.9 s,T=873~1 073 K,p=0.1 MPa,nN2∶n燃料=99.6∶0.4。根据1 000 K下生成量的多少,中间产物依次为C2H4、C3H6、CH4、1-C4H8、1,3-丁二烯(1,3-C4H6)和乙烷(C2H6)。前4种化合物占据了燃料的大部分碳,1,3-C4H6和C2H6来自以上主要中间产物的二次反应。
需要指出的是,图2及之后流动管反应器系统的模拟(图4和图8)时间均有不同程度的平移,其中,正辛烷的氧化反应模拟中,本模型模拟时间向左平移5 ms,LLNL机理模拟时间向左平移30 ms;异辛烷的氧化反应模拟中,本模型和详细机理模拟时间均向左平移10 ms;正丁基苯的氧化反应模拟中,本模型模拟时间向左平移11 ms,详细机理向右平移10 ms。这是由于理想流动管反应器模拟的初始条件会产生初始化的扰动,但不影响反应的时间或空间梯度。平移反应时间或空间是消除初始化扰动最简单有效的方式,且无其他影响,故在流动管反应器的模拟中适当平移可以使燃料转化与实验值匹配[17]。相对于LLNL详细机理,本简化模型预测值与实验值均吻合良好。对于图3的热解数据,LLNL机理的预测值在实验测量误差范围内,而本简化模型的预测值更接近实验测量值。

(a)n-C8H18
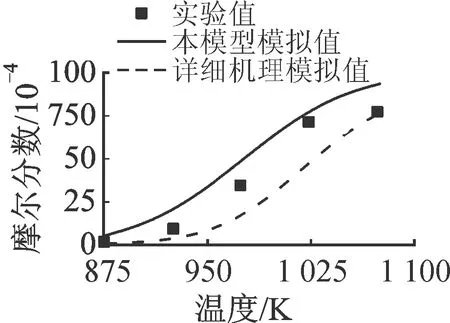
(a)C2H4
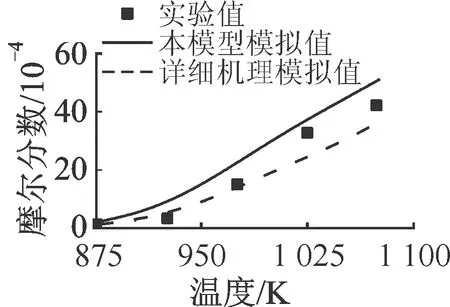
2.1.2 异辛烷 图4和图5分别对比了异辛烷(i-C8H18)氧化反应和热解反应主要中间产物的实验值与模拟值。与正辛烷类似,异辛烷氧化反应和热解反应实验数据分别来自流动管[15]和射流反应器系统[16],其中氧化反应具体条件为φ=0.99,T=1 080 K,p=0.1 MPa,nN2∶nO2∶n燃料=66∶1.2∶0.095;热解反应具体条件为τ=0.9 s,T=873~1 073 K,p=0.1 MPa,nN2∶n燃料=99.6∶0.4。相对于正辛烷,异辛烷转化过程的主要中间产物为异丁烯(i-C4H8)、C3H6、CH4和C2H4(C2H6为以上产物的二次反应产物)。该主要中间产物的不同可能导致正辛烷和异辛烷宏观燃烧特性不同,包括点火延时时间、层流燃烧速率等。
本文简化模型与LLNL详细机理对氧化反应中燃料转化和产物生成的预测十分接近,均较好复现了i-C4H8和C3H6,而对CH4和C2H4的预测稍显不足。对于热解反应,两类反应模型对CH4的预测差别较大,分别稍高和稍低于实验值;而对其他产物类似,均较好地预测了其生成和消耗趋势。
2.1.3 甲基环己烷 图6和图7分别对比了甲基环己烷(CH3cyC6)氧化反应和热解反应主要中间产物的实验值与模拟值。氧化和热解反应数据均来自流动管反应器,其中氧化反应的具体条件为φ=1.3,T=1 161 K,p=0.1 MPa,燃料摩尔分数为0.181 5%;热解反应具体条件为T=1 155 K,p=0.1 MPa,燃料摩尔分数为0.166 4%,O2摩尔分数为0.018%。由于环状结构的出现,甲基环己烷氧化和热解反应的主要中间产物,除了CH4、C2H4、C3H6等C1~C3组分,1,3-C4H6代替丁烯异构物(C4H8)成为来自燃料直接转化的主要中间产物(而非来自其他产物的二次反应),同时产生异戊二烯(i-C5H8)。
由图6和图7可见,本简化模型与LLNL详细机理对部分产物的预测值差别较大。具体地,本模型较好地预测了CH4、C2H4、1,3-C4H6和CO、CO2的生成或生成和消耗趋势,而对C3H6的预测值过低。相反,详细模型对C3H6的预测值则过高。对热解数据的预测,两类模型呈现类似的趋势。

(a)i-C8H18
(a)CH4
2.1.4 正丁基苯 图8和图9分别对比了正丁基苯(A1C4H9)氧化和热解反应实验值与模拟值。氧化和热解反应数据均来自流动管反应器,其中氧化反应的具体条件为φ=0.98,T=1 069 K,p=0.1 MPa,燃料摩尔分数为0.062%,O2摩尔分数为0.85%;热解反应的具体条件为T=985~1 226 K,p=0.003 947 MPa,nN2∶n燃料=99∶1。由图可见,由于苯环结构的出现,正丁基苯的转化除了生成CH4、C2H4、C3H6等C1~C3组分,也生成另外3类芳香烃物质甲苯(A1CH3)、苯乙烯(A1C2H3)和乙苯(A1C2H5),取代C1~C4物质成为主要中间产物。

(a)CH3cyC6

(a)CH3cyC6

(a)A1C4H9
图9显示,两种模型对热解反应过程的燃料消耗及中间产物浓度变化的预测接近且整体与实验值吻合良好。而由图8可见,对于氧化反应,本文模型对燃料消耗的预测过慢,LLNL详细机理的预测则过快;中间产物预测方面,本模型对CH4、C2H4、C3H6等小分子物质的生成和消耗过程,以及A1C2H5、A1C2H3等大分子物质的生成过程预测良好,而对C6H6、A1CH3的预测过低。相对地,详细机理除对C2H4和C6H6的预测过高,总体对A1C4H9的氧化过程模拟优于简化模型。这说明本模型对USC-Mech-Ⅱ模型添加的A1C2H5、A1C2H3等芳香烃的化学反应依然不够完全。

(a)A1C4H9
2.2 燃料热解产物的碳分布
为探究4类燃料的中间产物分布对整个燃烧过程的影响,本节考察了各主要中间产物的碳分布,即中间产物含碳量与燃料转化碳量的比值。图10为4类燃料在燃料转化率为50%时主要中间产物的碳分布。由图可见,氧化和热解反应的碳分布几乎相同,说明氧气对中间产物的碳分布影响较小;在燃料消耗尽之前,其作用主要是增加反应系统活性,提升反应系统整体转化速率。

(a)n-C8H18
4类燃料的纵向比较显示,正辛烷、异辛烷最主要的中间产物是C1~C4中的烯烃(占燃料转化碳含量的90%以上)。其中,正辛烷以C2H4为主,其次为C3H6和1-C4H8;异辛烷以1,3-C4H8为主,其次为C3H6。甲基环己烷的主要中间产物仍然是C1~C4,其中以C2H4和1,3-C4H6为主,但由于加入了环结构,还产生了1,3-C5H8、i-C5H8以及环己烯(C6H10);由于正丁基苯含有芳烃环,A1C2H5和A1C2H3等成为主要的中间产物,C1~C4中仅C2H4作为主要中间产物。这也说明正丁基苯中间产物模拟出现偏差的主要原因是基础燃料模型对芳香烃动力学描述不完善。如2.4和2.5节所述,以上含不同官能团的4类燃料主要中间产物的不同将导致不同的宏观燃烧特性。
2.3 热解模型分析
大分子燃料的高温热解简化模型包含7个总包反应,分别为1个C—C键离解反应(R1)以及6个脱氢反应,即燃料+H·(R2),燃料+CH3·(R3),燃料+OH·(R4),燃料+HO2·(R5),燃料+O·(R6),燃料+O2(R7)。为探究以上各反应对燃料转化的影响,本节通过敏感性系数和相对燃料消耗率(即每个反应消耗速率与总消耗速率之比)比较了R1~R7在燃料消耗中的作用。
图11比较了4类燃料在50%转化率时反应R1~R7的敏感性系数和相对燃料消耗率。除了甲基环己烷中R3是最重要的反应外,其他3类燃料最重要的反应均是R1,其次是R2或R3。正辛烷的相对燃料消耗率由大到小依次是R2、R1、R3和R4~R7,甲基环己烷为R2、R3、R4、R1和其他,而异辛烷和正丁基苯中则为R1、R2、R3和R4~R7。

(a)n-C8H18
综上,R1,即燃料C—C键离解反应在燃料转化中作用最显著,不仅作为起始反应,产生最初的H·、CH3·等活性自由基,而且是燃料消耗的主要方式。R2和R3,即H·和CH3·参与的燃料脱氢反应也对燃料消耗有很大贡献,而含氧自由基的影响则相对有限。
2.4 点火延时时间
图12比较了4类燃料的点火延时时间实验值和模拟值。数据均来自激波管,具体实验条件为:对于正辛烷[22],燃料+4% O2+Ar,φ=0.98,p=0.2 MPa;对于异辛烷[23]和甲基环己烷[24],燃料+4% O2+Ar,φ=0.98,p=0.15 MPa;对于正丁基苯[25],燃料+空气,φ=0.98,p=0.1 MPa。总体而言,本简化模型和LLNL详细机理的预测值均与实验值吻合良好。
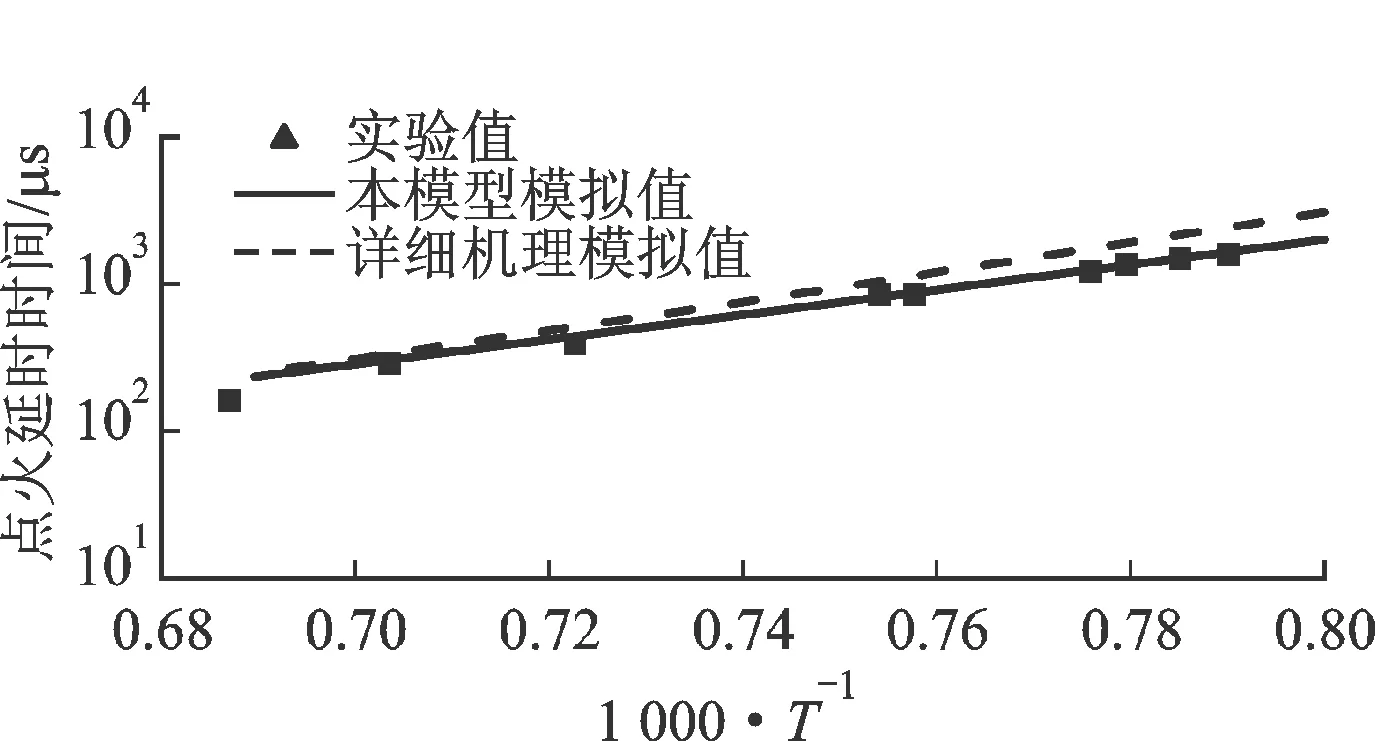
(a)n-C8H18
4类燃料的纵向对比显示,相同实验条件下(注:正丁基苯在空气中,其他3类燃料在4% O2+Ar中),点火延时时间从小到大依次为正辛烷、甲基环己烷、异辛烷和正丁基苯。结合2.2节的碳分布比较,正辛烷的中间产物以C2H4为主、甲基环己烷以C2H4和1,3-C4H6为主、异辛烷以i-C4H8为主;而C2H4、C4H6、i-C4H8的点火延时时间依次增加[19-21],即各主要中间产物的活性依次降低,从而导致其母燃料类似的点火特性。
2.5 层流燃烧速率的验证
4类燃料的层流燃烧速率实验值和模拟值的比较如图13所示。具体实验条件为:对于正辛烷[27-28],T=353 K,p=0.1,0.2 MPa;对于异辛烷[29-31],p=0.1 MPa,T=298,353 K;对于甲基环己烷[26],p=0.1 MPa,T=353,403 K;对于正丁基苯[14],p=0.1 MPa,T=358,398 K。本小节对正辛烷、异辛烷和正丁基苯详细机理的层流燃烧速率验证,来自LLNL或更新过的LLNL详细机理,而甲基环己烷来自JetSurF v1.1机理[26](LLNL详细机理缺少甲基环己烷反应体系的物质输运参数)。如图13所示,除本简化模型对正丁基苯在稀燃区的预测处于实验误差下边缘外,两类模型对所有燃料在不同实验条件下的预测值均与实验值吻合良好。正丁基苯出现该模拟结果的原因可追溯至基础燃料模型USC-Mech-Ⅱ缺少对其热解产物主要中间产物(A1C2H5、A1C2H3等)的动力学描述,而这也正说明燃料的宏观燃烧特性主要由中间产物的燃烧化学反应特性决定。
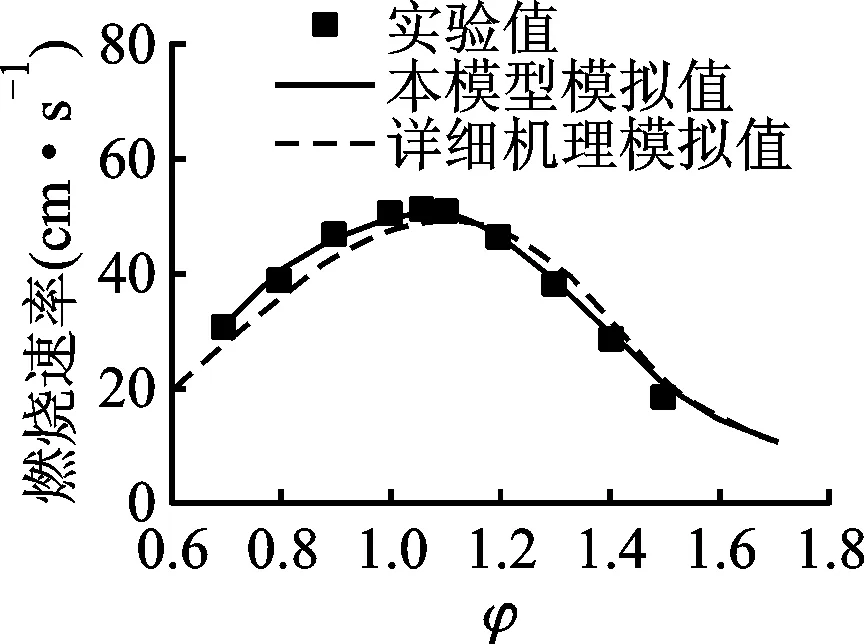
(a)n-C8H18,353 K,0.1 MPa
2.6 模型简化
以上述已建立好的n-C8H18简化模型为例,采用误差传播直接关系图谱法对其进一步简化。原始简化模型包含124个化学组分和854个基元反应,简化后,化学反应和基元反应的数量分别为56个和387个。显然,结合其他简化方法,简化后的模型规模或可进一步缩小。由图14可见,模型简化前后,对点火延时时间和层流燃烧速率的预测吻合良好。该简化模型的规模可与先进计算流体力学CFD软件或代码有效耦合,用于对真实发动机系统的仿真模拟。

(a)模型简化前后点火延时时间对比
3 结 论
本文建立了正辛烷、异辛烷、甲基环己烷和正丁基苯等含4类官能团的大分子碳氢燃料的简化动力学模型。经与基础燃烧反应动力学数据,包括氧化反应和热解反应重要中间产物分布、点火延时时间、层流燃烧速率等的对比,显示本简化建模方法不仅对直链烷烃和支链烷烃有较好描述,同时也适用于环烷烃和烷基芳香烃的燃烧化学。该简化模型仅包含124个化学组分和854个基元反应,可进一步缩减。以n-C8H18为例,进一步简化后,化学反应和基元反应的数量分别为56和387个。为进一步扩展该方法的使用范围,下一步研究将探究该类简化建模方法扩展至较低温度区域(包括负反应温度系数区域,即NTC区域)的情况。
