助催化剂对烯烃配位聚合反应的影响*
2021-02-12王雪儿傅智盛范志强
王雪儿,傅智盛,范志强
(浙江大学高分子科学与工程学系,浙江 杭州 301127)
聚烯烃是应用十分广泛的高分子材料,自Ziegler-Natta催化剂问世以来,烯烃配位聚合催化剂及其催化机理的研究得到了蓬勃发展。设计合成新的催化剂和调控聚合条件可以对聚合物的结构进行控制,从而获得性能优异、品类繁多的聚烯烃材料。绝大多数烯烃聚合催化剂都需要在助催化剂的作用下才能引发烯烃聚合,并且助催化剂的成本时常高于催化剂前体。从经济角度而言,了解助催化剂在聚合过程中的作用,对烯烃配位聚合的基础研究和工业生产技术的发展都具有十分重要的意义。
大量研究表明,在过渡金属催化烯烃聚合反应过程中,助催化剂对催化剂的催化活性、聚合动力学行为以及聚合产物的相对分子质量和立构规整度等方面都有重要的影响,其重要性可归结为:首先催化剂前体必须经过合适且高效的助催化剂活化才能成为聚合反应中真正的活性中心;其次一个成功的活化过程要考虑助催化剂的Lewis酸性、与催化剂结构的适配度等因素;最后助催化剂在活化后成为催化剂活性中心阴阳离子对的组成部分,可能显著影响聚合的动力学过程和聚合物的结构性能[1]。
1 助催化剂的主要类型
1.1 烷基铝
烷基铝及氯化烷基铝是烯烃配位聚合催化体系的重要组分。1953年Ziegler发现TiCl4和三乙基铝(AlEt3)双组分催化体系可以使乙烯在温和的条件下发生聚合反应[2]。次年Natta发现TiCl3和一氯二乙基铝(AlEt2Cl)催化体系可以制备全同或间同聚丙烯,并且首次提出了烯烃定向聚合的概念[3]。钒系催化剂在AlEt2Cl的作用下催化乙烯、丙烯共聚可以获得序列分布较均匀的共聚物,这类催化剂还可以在低温下(-60 ℃)催化丙烯聚合得到相对分子质量分布相当窄的间规聚丙烯[4]。图1为钛系Ziegler-Natta催化剂由烷基铝活化成为活性中心的过程,在这个反应中,烷基铝作为烷基化试剂和还原剂将催化剂前体烷基化为带有一个烷基配位及一个正电荷的活性中心,从而催化乙烯进行聚合反应[5]。
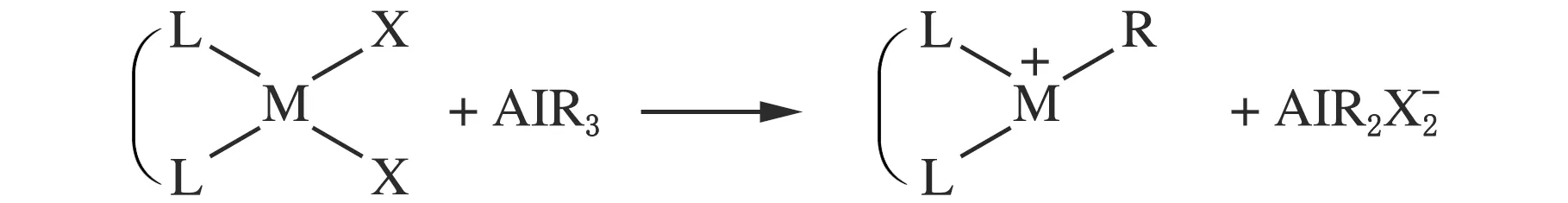
图1 烷基铝活化机理
1.2 甲基铝氧烷(MAO)
Sinn等[6]首次于1980年用三甲基铝(TMA)水解的方法合成了MAO,以MAO作为茂金属催化剂的助催化剂,可以使茂金属催化剂具有很高的催化活性,相比烷基铝作助催化剂提高了几个数量级。一种广泛被接受的理论是MAO是由三维笼状结构的(AlOMe)n和残留的TMA组成。MAO的组成受制备方法的影响,很难用实验方法测定两种组分的含量。
1.3 有机硼化合物
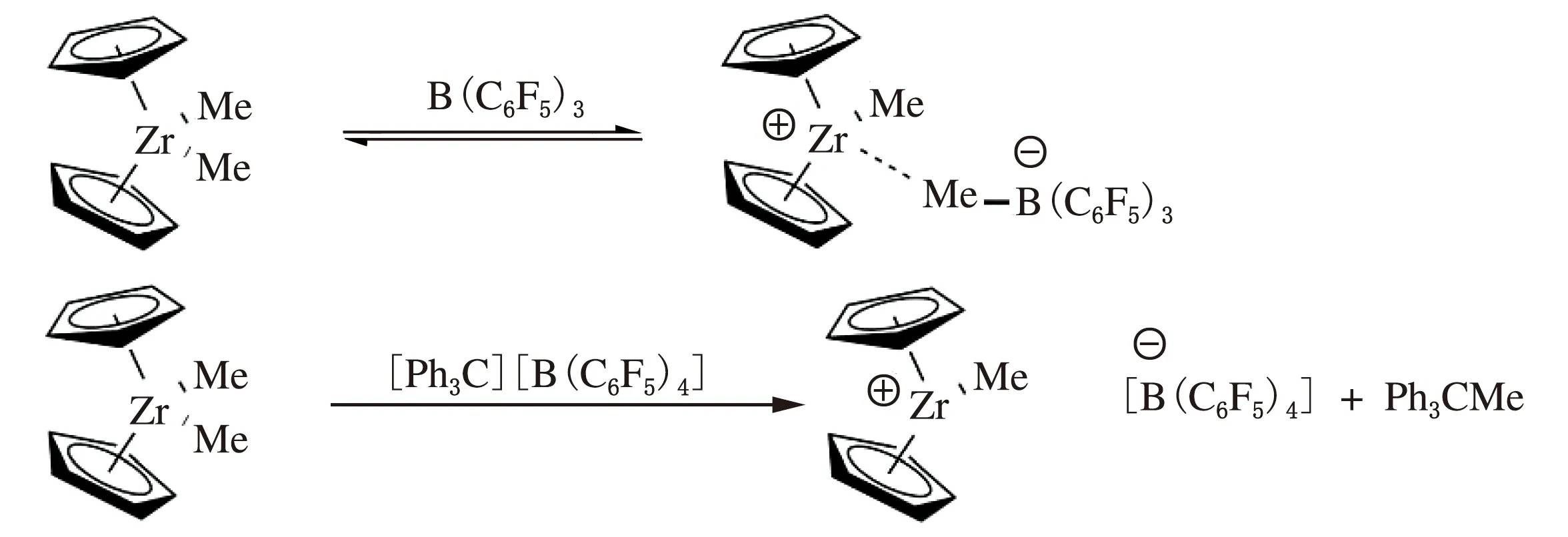
1991年,Marks[7]和Ewen[8]的团队分别首次将B(C6F5)3(FAB)作为烯烃聚合催化剂的助催化剂,FAB和二甲基二茂锆催化剂配合后在常温常压下催化乙烯聚合,催化活性可达4.5×106g/(mol·h)。这种具有大体积配位阴离子的有机硼化合物可以替代MAO活化茂金属催化剂,在硼化合物的作用下,茂金属催化剂前体发生离子化而成为烯烃聚合的阳离子活性中心。硼化合物与MAO相比有两个明显的优势:一是只需等摩尔的硼化合物就可以活化茂金属催化剂,并且对烯烃聚合有很好的催化活性;二是活化后生成的离子对有确定的结构,研究者们可以更好地通过实验研究催化剂前体和助催化剂之间的相互作用[9]。
2 助催化剂对茂金属催化体系的影响
2.1 活化机理
茂金属催化体系的活性中心通常由过渡金属阳离子和非配位阴离子组成,这类活性中心可以由中性的茂金属催化剂前体和助催化剂原位生成。
2.1.1 MAO活化机理


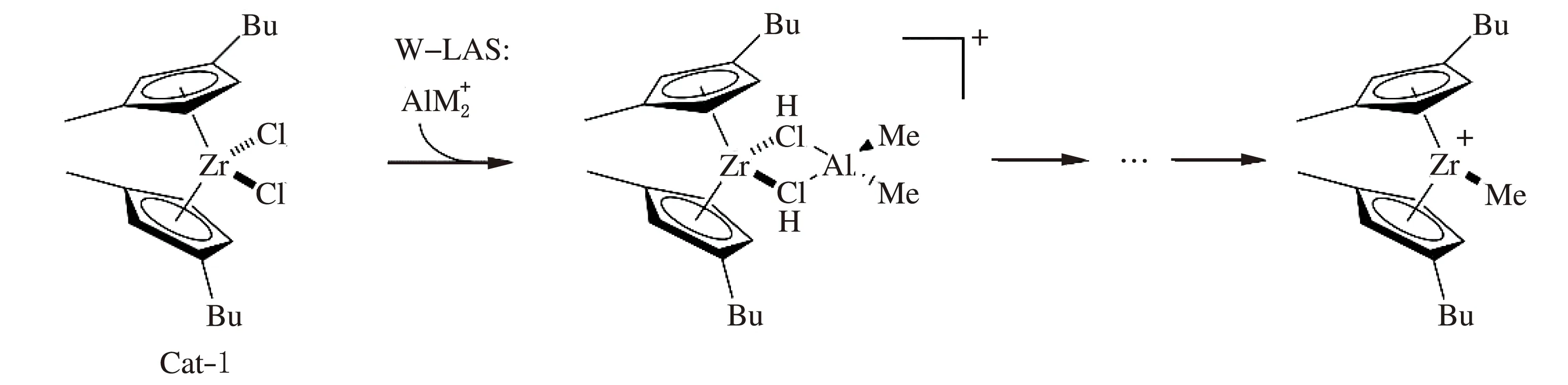
图2 Cat-1/MAO活化过程示意图
2.1.2 有机硼化合物活化机理
Ⅱ.由PhMe2NH+发生质子分解消除甲基
Ⅰ.由中性Lewis酸或Ph3C+消除甲基
Nakashima等[11]报道了上述三种硼化合物对二甲基二茂锆催化乙烯聚合的影响。其中TTB的乙烯催化活性是三种硼化合物中最高的。这是由于FAB进行烷基消除生成的[MeB(C6F5)3]-中Me和金属存在agostic作用(图4左)。而对于AB,副产物PhNMe2可以捕获金属阳离子并与其配位(图4右),这两种相互作用都会影响催化活性。
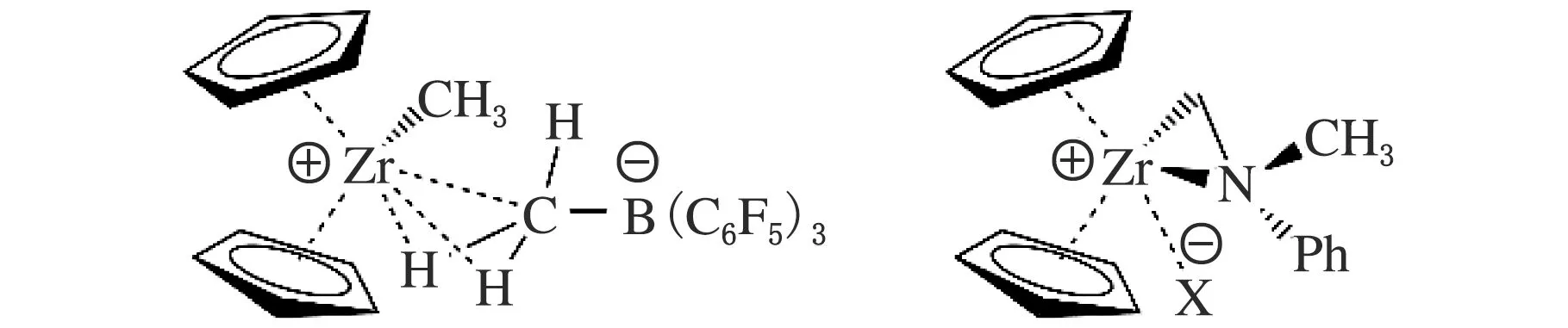
图4 两种离子对相互作用[1]
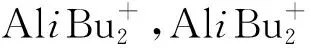
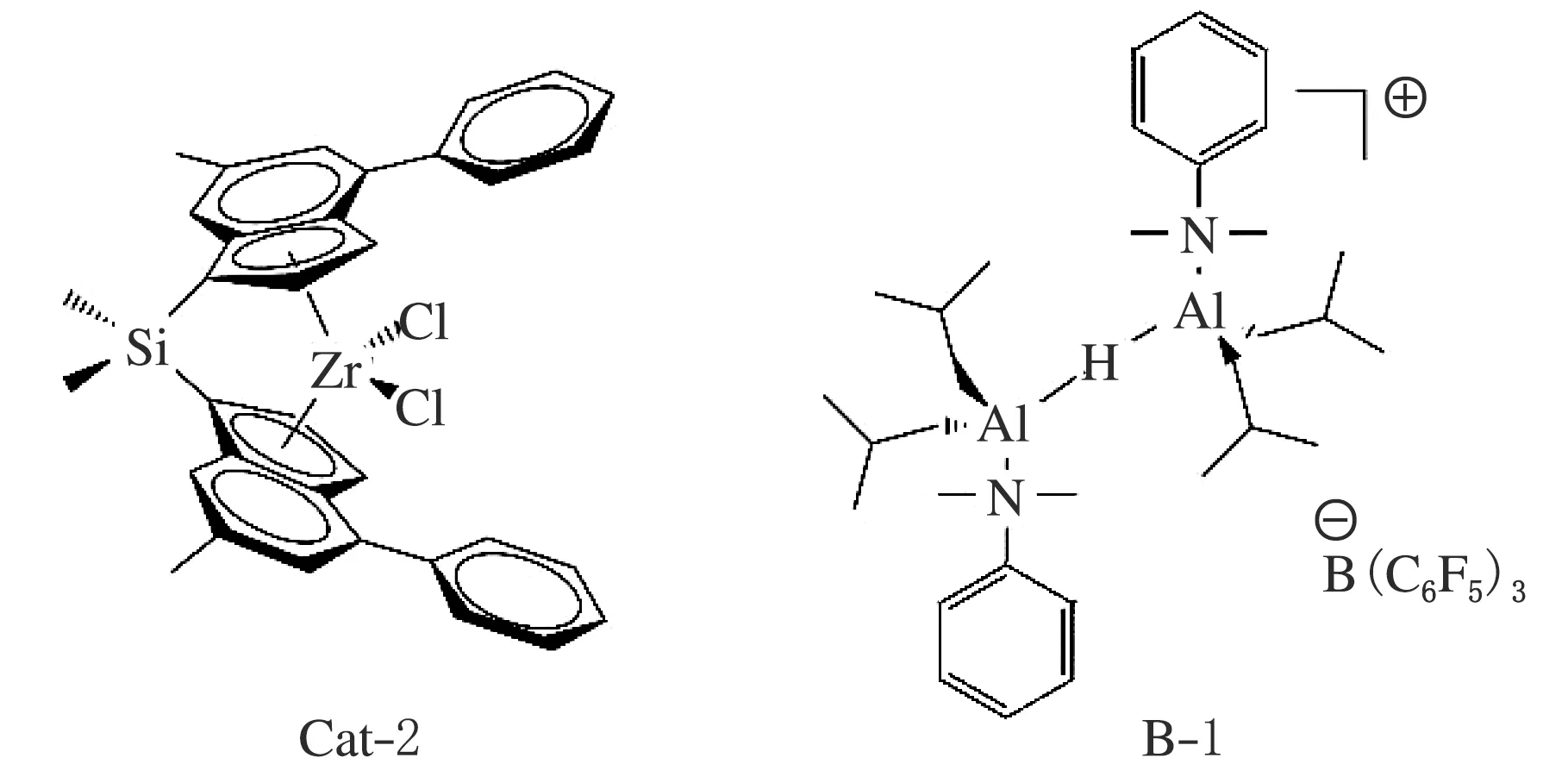
图5 Cat-2和B-1的结构示意图[13]
2.2 结构-性能关系
从各类助催化剂的活化过程来看,不同结构的催化剂和助催化剂之间的活化机理不同,所形成的聚合活性中心的结构也不同。对于茂金属催化剂与助催化剂形成的离子对而言,阴离子会与单体竞争配位,离子对的形成、形态、浓度以及溶剂化作用都对聚合反应的催化活性、化学选择性、区域选择性和立构选择性有至关重要的影响。
2.2.1 离子对的形态结构特点
烯烃配位聚合反应所使用的溶剂往往是低极性的烃类溶剂,因此溶液中很难存在自由离子,聚合反应活性中心通常以离子对或离子对聚集体的形式存在。由过渡金属参与的离子对可分为两种类型:一种是内层离子对,其阴离子和金属中心间存在弱静电作用;另一种是外层离子对。[Cp2ZrMe]+[MeB(C6F5)3]-和[Cp2ZrMe]+[B(C6F5)4]-都属于内层离子对,向溶液中加入另一种Lewis酸[四氢呋喃(THF)或三苯基膦(PPh3)],THF或PPh3会参与金属中心的配位[13],使离子对转变为外层离子对,这个过程类似于烯烃单体与金属中心配位插入的过程。
对于典型的茂金属催化剂而言,阴离子的结构的影响是显著的。非配位阴离子最好有较大的体积位阻,带一个负电荷并且电荷在整个分子内高度离域[14]。将FAB中的全氟苯基替换为体积更大、电荷离域程度更高的联苯基(图6中的PBB和BPB)或萘基(图6中的PNB),可以提高茂金属催化剂特别是限制几何构型茂金属催化剂Cat-3(图7)的催化活性[15-17]。二硼烷B-2(图6)与Cat-4(图7)配合可以有效催化乙烯和异丁烯、亚甲基环戊烷和亚甲基环己烷等空间位阻较大的共单体发生共聚,共聚单体的插入率比单核钛催化剂提高了两倍以上[18]。Marks等合成了B-3(图6),由于刚性萘环的限制,硼上的—C6F5与萘环处于互相垂直的位置,使硼具有很强的缺电子性,与二甲基二茂锆配合后的催化活性比FAB提高了一个数量级[19]。
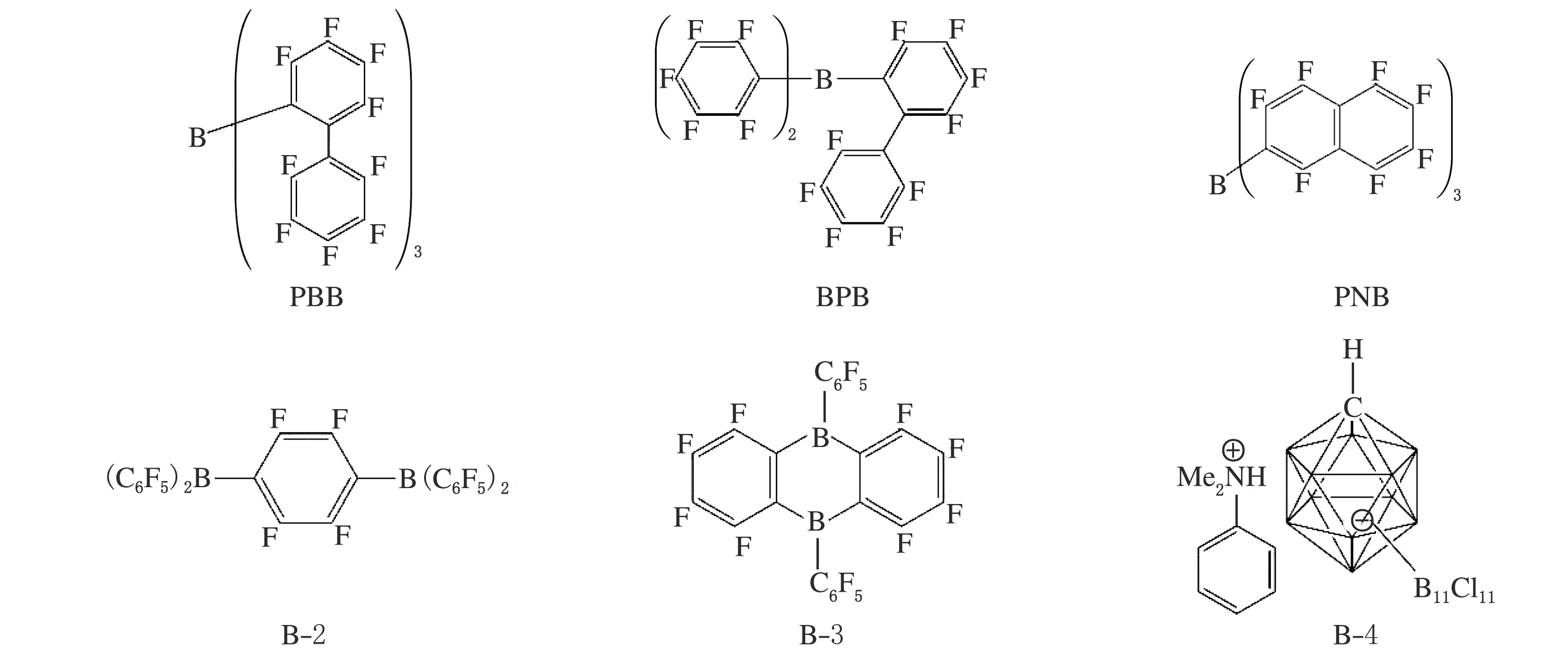
图6 硼化合物结构示意图
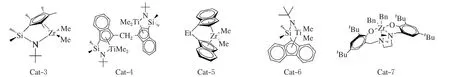
图7 Cat-3~Cat-7结构示意图[11].[16-19].[21].[24]
Metz等[20]最先报道了Cat-5/TTB体系(图7)在催化丙烯等规聚合中的应用,该催化体系在20 ℃下的催化活性是Cat-5/MAO体系的6倍。[B(C6F5)4]-与金属中心的相互作用比[MeB(C6F5)3]-与金属中心的相互作用更弱,更有利于单体配位插入,因此TTB对丙烯的催化活性比FAB高出许多,获得聚丙烯的等规度更高。Chien等[21]报道了Cat-6/TTB体系(图7)可以催化降冰片烯和长链α-烯烃共聚得到梯度共聚物,共聚物的数均分子质量超过了105g/mol,平均分子量分布在1.3以下。对TTB进行结构修饰可以改变其阴离子结构,例如将—C6F5对位上的氟用—SiR3取代,改善了TTB在低极性溶剂中的溶解性。
对于AB而言,铵基阳离子上大位阻的苯基有利于减弱其与金属中心的作用,比如一些位阻较小的Me3N+和NH4+容易与金属中心发生配位,使得催化剂对烯烃聚合的活性较低。在铵基阳离子或三苯基碳阳离子上修饰长链烷基可以改善硼酸盐的溶解性。Yuan等[22]合成了一种不同阴离子的铵基硼酸盐B-4,B-4与Cat-3催化剂配合后对乙烯的催化活性与AB相近,特别的是它在高温下(190 ℃)较稳定,且阴离子上的C—H可以用来引入不同的功能基团,从而设计出更多新型硼酸盐助催化剂。
Gunther等[23]制备了一种中性氟芳基硼烷B-5(图8),它在碱的存在下转化为硼烷阴离子,尽管其Lewis酸性低于FAB,但它具有较好的溶解性,和Cat-6配合后对丙烯在甲苯和庚烷中的聚合有较好的催化性能,可以催化丙烯聚合得到超高分子量(>106g/mol)的聚丙烯。相较于Cat-6/TTB体系,Cat-6/B-5体系催化活性较低,引发效率低,链转移发生较少,聚合产物的相对分子质量较高。FAB也能溶于庚烷中,但由于形成的阴阳离子对在庚烷中较为紧密导致丙烯聚合活性很低。Takeshi等研究了B-5活化二甲基二茂锆的过程,认为B-5在这个过程中同时作为Lewis酸和Brønsted酸。首先B-5作为Lewis酸与锆金属中心作用发生甲基消除反应,反应较长时间后,甲基锆发生缓慢的质子化反应生成硼蒽阴离子。B-5比FAB具有更好的活化性能归因于这一步质子化反应,生成了结构更松散的离子对。基于这一特点B-5及其衍生物有望进一步用作稳定各种金属阳离子中心的非配位阴离子。

图8 二甲基二茂锆催化剂/B-5活化过程示意图[11]
通过改变阳离子配体的空间位阻和电子效应可以对离子对的形态进行调整,对催化剂的区域选择性和立构选择性也有重大影响。Kol等[24]合成了以二胺-二苯氧基(ONNO)为配体的锆催化剂Cat-7(图7),Cat-7对1-己烯有很好的聚合活性。用FAB活化Cat-7在室温下催化1-己烯的聚合活性可以达到18 kg/(mol·h),可以得到等规度大于95%的聚己烯,将配体苯环上的tBu替换为甲基后,得到的聚合物等规度下降至50%。Clancaleoni等[25]将Cat-7苯环对位上的tBu替换为甲氧基后合成了Cat-8,他们探讨了FAB和TTB对Cat-8催化丙烯聚合的影响,研究发现催化活性并不受阴离子结构的影响。原因在于阳离子配体的特殊性,Cat-8与FAB和TTB反应形成的离子对均是外层离子对。进一步的研究表明,溶液中该催化剂存在面式-面式(mer-mer)(图9左)和经式-经式(fac-fac)(图9右)两种构型,mer-mer构型比较稳定,但由于其金属烷基键与空位处于相对的位置而几乎没有聚合活性。fac-fac构型为聚合反应发生时的构型,由于阴离子不参与构型转变过程,因此不同的助催化剂对催化活性几乎没有影响。
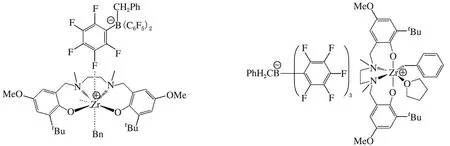
图9 Cat-8被FAB活化后的两种离子对异构体
2.2.2 离子对的自聚集与溶剂化作用
由于阴阳离子间的弱相互作用,离子对在溶液中会发生自聚集现象。研究表明离子对越松散、溶剂的极性越低,越容易发生离子对的自聚集。目前离子对的自聚集对聚合反应的影响尚不明确,Brintzinger和Bochmann等认为离子对的自聚集对聚合反应的动力学过程有很大影响,例如提高TTB的浓度会使催化活性得到提高,他们认为这一现象正是由于浓度的增大使得离子对自聚集更容易发生而导致的。Marks和Piers对此持不同观点。他们认为自聚集现象并不会影响聚合的动力学过程[26]。
溶剂化作用在调节聚合行为方面起着重要的作用。为了更好地研究溶剂化作用的影响,Gao等[27]在Ph3C+苯基的对位上修饰以-n-octyl来改善TTB的溶解性,该硼酸盐与催化剂Cat-9(图10)配合后,在甲苯和甲基环己烷(MeCy)中的乙烯聚合结果有明显的差异。甲苯作溶剂时得到低相对分子质量、低支化度的聚乙烯,而甲基环己烷作溶剂时得到低相对分子质量高支化度的聚乙烯(HBPE),聚合活性也比甲苯作溶剂时高出两倍。他们认为这一差异是溶剂化作用导致的。在聚合过程中,活性中心与生长的聚合物链发生β-H消除,重新产生空位和端烯基大分子单体,对于极性较高的甲苯溶液,甲苯分子更易和锆金属中心发生配位,阻碍了比它空间位阻更大的大分子单体的重新插入,因此产物的支化度较低。
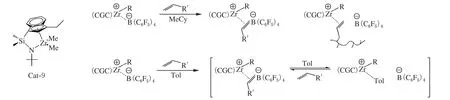
图10 甲基环己烷和甲苯溶液中离子对的形态差异
2.2.3 浓度的影响
离子对的形态和催化行为还受到离子对浓度和助催化剂比例的影响。Xiong等[28]报道了Cat-10(图11)在不同[B]/[Zr]比下催化1-己烯聚合的实验结果。

对于MAO而言,助催化剂浓度的影响更显著,活化反应往往需要很高的[Al]/[M]比。在较低[Al]/[M]比(<100)时,MAO的活化反应是不完全的,生成[LnMMe]+[MeMAO]-(ISIP)和二聚体络合物[LnMMe(μ-Me)LnMMe]+[MeMAO]-,这两者都被认为不具有催化活性。在高[Al]/[M]比(>100)时生成真正的活性中心[LnMMe]+[MeMAO]-(OSIP)。也有研究者认为过量的MAO可以在溶液中形成更大的(AlOMe)n笼的聚集体,使电荷离域程度更高来减弱离子对相互作用。催化剂前体的甲基或卤素配体也能影响阴离子的配位能力,LnMCl2比LnMMe2需要更多的MAO来活化,这或许是因为Cl—MAO-比Me—MAO-与[LnMMe]+形成的离子对更紧密所导致的。
3 助催化剂对后过渡金属镍/钯催化体系的影响
自Brookhart对(α-二亚胺)镍/钯催化剂的开创性研究以来,后过渡金属催化剂就引起了人们广泛的关注[29]。相较于Ziegler-Natta催化剂和均相茂金属催化剂,后过渡金属催化剂具有较好的极性基团耐受性、较高的催化活性和链转移速率等特点。
3.1 助催化剂对聚合物结构性能的影响
MAO、改性的甲基铝氧烷(MMAO)、AlEt2Cl和三氯三乙基二铝(Al2Et3Cl3是后过渡金属镍/钯催化剂最常用的助催化剂。研究表明,与MAO或MMAO相比,由AlEt2Cl或Al2Et3Cl3活化的后过渡金属镍催化剂具有更高的催化活性,获得的聚乙烯产物具有更高的支化度、更低的熔点和相对分子质量[30-35]。助催化剂中的Cl原子可以与镍作用形成Cl-Ni桥,Cl的吸电子效应促进了链增长及链转移反应的发生。在研究助催化剂的作用时,应考虑催化剂配体的结构。例如一些带有亚胺吡啶配体的镍催化剂在MAO活化下表现出比AlEt2Cl、AlMe2Cl活化时更高的催化活性[36]。就烷基铝的浓度而言,[Al]/[Ni]比通常在一定程度上影响催化活性和聚合产物的相对分子质量。随着[Al]/[Ni]比的增大,催化活性在一定范围内达到最大值,由于链转移速度加快,产物的相对分子质量随[Al]/[Ni]比的增大而减小,相对分子质量分布变窄。烷基铝的种类浓度一般对聚合物的拓扑结构影响不大[37]。
Jian等[38]用聚乙二醇链段修饰(α-二亚胺)镍催化剂的骨架结构,合成了“三明治”结构的催化剂Cat-11(图12),以MAO、MMAO和AlEt2Cl为助催化剂进行乙烯聚合实验。他们发现随着配体骨架上乙二醇结构单元的增加,催化活性增大,聚合产物的支化度明显下降(由77/1 000C变为19/1 000C)。Jian推测这是由于助催化剂的铝原子和聚乙二醇链段上的氧原子发生配位导致的,这种作用使助催化剂一定程度上被固定在金属中心周围,一方面使金属中心的缺电性增强,另一方面增加了金属中心周围的空间位阻,减少了链行走反应的发生。
烷基化的钯催化剂前体可以在硼酸盐的作用下引发烯烃单体聚合。Mu等[39]合成了以双磷单氧化物为配体的钯催化剂Cat-12(图12),以NaB[3,5-(CF3)2C6H3]4(NaBArF)为助催化剂,该体系可以催化乙烯和6-氯-1-己烯发生共聚,在0.5 MPa、80 ℃的聚合条件下催化活性可达106g/(mol·h),共聚物中6-氯-1-己烯的摩尔插入率为5.3 mol%。
一些具有特殊配体结构的催化剂可以在不加入助催化剂的情况下引发聚合反应。Chen等[40]报道了一系列含膦磺酸盐配体的单组份镍催化剂Cat-13(图12),该体系可以较好地催化乙烯和乙烯基三甲氧基硅烷、丙烯基醚及功能化降冰片烯等极性单体共聚,催化活性最高可达1.9×105g/(mol·h),共聚物的数均分子量最高可达4.46×105g/mol,极性单体的摩尔插入率为0.7%~7.6%。

图12 Cat-11~Cat-12结构示意图
3.2 活性中心结构
对(α-二亚胺)镍/钯催化剂的配体结构进行修饰可以提高催化剂的热稳定性,从而获得结构性能更好的聚合物,人们在这一方面的研究已经取得了相当多的成果。然而对于这类催化剂结构性能关系的基础理解还停留在现象阶段,通过实验方法来明确反应溶液中活性中心的结构仍存在很大的困难[41]。
Talsi等用1H-NMR和13C-NMR以及电子顺磁共振(EPR)的方法对助催化剂(MAO和MMAO)活化Cat-14(图13)的过程做了系统的研究。研究表明在-40 ℃下,Cat-14/MAO催化体系在反应初期,生成了双金属络合物离子对[LNiⅡ(μ-Me)2AlMe2]+[MeMAO]-。而在相同条件下,Cat-14/MMAO催化体系则生成了离子对[LNiⅡ-tBu]+[MeMMAO]-,这种二价镍络合物离子对只能在-40~-20 ℃下稳定存在[43],在室温以及更高的温度下会迅速转变为镍的一价络合物。起初Talsi认为镍的一价络合物的结构为[LNiⅠ(S)]+[MeMMAO]-(S为溶剂分子)[44]。他们对比研究了Cat-14/MMAO体系和Cat-14/AlMe3体系,将MMAO或AlMe3在25 ℃下预活化1 h,所得到的催化活性降低为原来的一半,两次聚合得到的聚合产物的数均相对分子质量及平均分子量分布并未有大的变化。EPR则证明了Cat-14/MMAO体系和Cat-14/AlMe3体系在[Al]/[M]比大于100时预活化后主要生成了结构相似的3MMAO和3AlMe3,不同的溶剂(甲苯、邻二氟苯、庚烷)对3MMAO的EPR参数影响不大。并且在Cat-14/AlMe3体系中,AlMe3的Lewis酸性较弱,并不容易从镍中心上消除甲基得到[LNiⅠ(S)]+[AlMe4]-,因而推翻了先前溶剂分子参与配位的假设,从而提出如图13中3MMAO和3AlMe3的结构,一价镍络合物3MMAO和3AlMe3仍是四配位结构,在MMAO和AlMe3的作用下,负电荷离域生成了非共价加合物[45]。
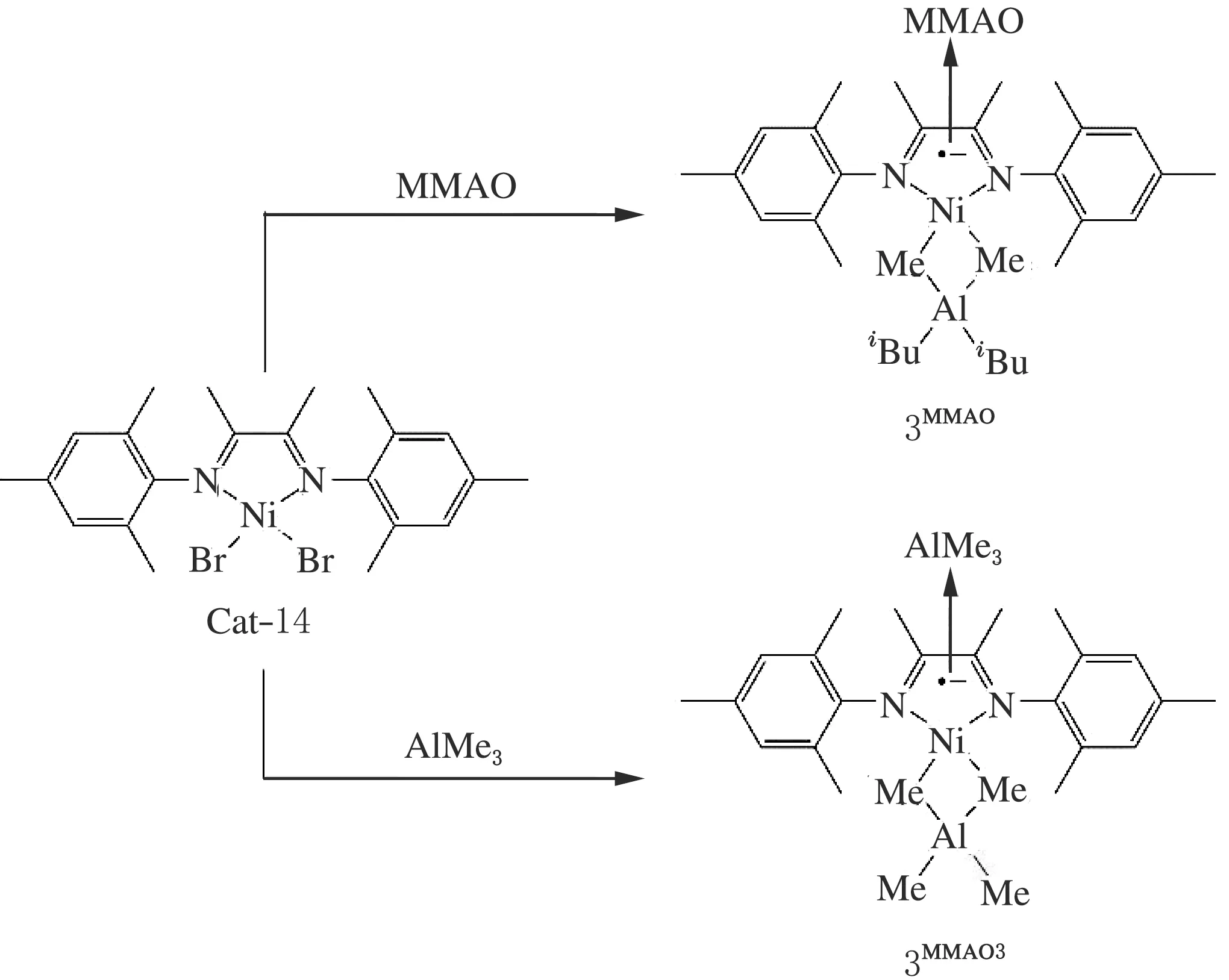
图13 3MMAO和3AlMe3的结构示意图
4 结 论
研究助催化剂在聚合反应中的作用对聚烯烃新材料的研发和工业化生产技术的发展具有十分重要的意义。传统的Ziegler-Natta催化剂在工业上的应用相当广泛,其助催化剂烷基铝的种类和加入量会对催化活性以及产品粒径产生影响。例如加入量过少会造成催化活性降低,加入量过高则会导致成本浪费,另外烷基铝与给电子体的相互作用也会影响聚丙烯产品的等规度。对于茂金属产品而言,茂金属聚烯烃、茂金属聚烯烃弹性体和新材料环聚烯烃合成用的茂金属催化剂都需要MAO作为助催化剂,但受制于技术和产品质量问题,MAO一直依赖于进口,价格昂贵,MAO国产化成为了限制国内茂金属产品产业化发展的关键因素。因此开发工业合成难度较低、经济成本低的新型助催化剂具有广阔的应用前景,研究各类助催化剂活化机理、活性中心结构和产物性能之间的关系可以为新型助催化剂的结构设计提供重要指导意义,也为原有催化剂的催化效能的改善提供更多可能。
助催化剂一般通过与催化剂前体反应使其转变为阳离子络合物从而引发聚合反应,继而作为该阳离子的反离子留在体系中。除了作为清除剂外,在后过渡金属镍/钯催化体系中,助催化剂还可以作为链转移剂存在。在均相茂金属催化体系中,MAO做助催化剂可以获得相当高的催化活性。硼化合物作为MAO的替代物为人们研究活性中心离子对的结构提供了支撑,不同结构的催化剂和硼化合物配合可以调整离子对的形态结构,进而对聚合物的立构选择性、区域选择性以及共聚单体的插入产生影响。助催化剂的浓度和溶剂化作用会显著影响聚合反应过程。在后过渡金属镍/钯催化体系中,不同助催化剂往往影响催化活性和产物的相对分子质量,而聚合活性中心的真正结构还有待进一步研究。
