细菌的泛基因组分析
2021-01-19关海霞
关海霞
天津大学理学院,天津300072
过去40 年中,基因测序技术不断更新发展。最早提出以双脱氧核苷酸末端终止法为代表的第一代测序技术推动了基因组学的建立。随后不断地开发出第二代测序技术(以Roche 公司的545 测序技术、ABI 公司的Solid 测序技术及illumina 公司的Hiseq和Solexa 技术为代表)及以单分子测序为特点的第三代测序技术。
DNA 测序技术的发展推动了生物学研究进入一个崭新的层面[1]。研究人员发现,对某一物种进行研究时,选取其中的一株菌株为研究对象不能完全反映该物种基因组的特征,而泛基因组分析方法的提出解决了这个难题[2]。本文就泛基因组的研究策略、模型构建及其在细菌中的研究应用作一综述。
1 泛基因组概念
2005 年,泛基因组(pan-genome)概念被首次提出[3]。泛基因组表示某一物种全部基因的总称,包括核心基因(core genome)及非必需基因(dispensable genome)。核心基因组是存在于所有菌株中的基因,管家基因是其重要的组成部分,参与基础生物学过程,如基因表达、能量转换、氨基酸代谢等。非必需基因由附属基因(accessory gene)和特异性基因(unique gene)两部分构成[3-4]。附属基因指存在于2株或多株菌株中编码次级代谢途径相关的基因;菌株特异性基因指某株菌株中独特存在的基因,是通过基因水平转移(horizontal gene transfer,HGT)的方式获得,与该菌株的特定性状相关,如产抗生素、耐受性、毒力及环境适应性等[5]。
维恩图是泛基因组可视化的普遍形式,不同圆圈分别代表给定不同菌株的基因组,之间的重叠和非重叠部分分别代表该集合菌株的泛基因组的组分,即核心基因组、附属基因及特异性基因,见图1。

图1 泛基因组成的维恩图Fig.1 Venn diagram of pan-genome
泛基因组模型可提供基因组异质性的信息(基因含量)。泛基因组的拟合模型可用于推测:①特定集合菌株的全基因组即泛基因组的大小;②该集合核心基因组的大小;③每个新基因组测序分析平均增加的新基因数目,其与泛基因组的类型(开放/ 闭合)有关。对于闭合的泛基因组(close pan-genome),新菌株的基因组测序完成产生新基因的可能很小,物种的泛基因组大小增加到一定程度后会收敛于某一值附近;而对于开放的泛基因组(open pangenome),每增1 株新菌株通常有新的基因加入到物种的基因库中,泛基因组也会随之不断增大[2,6]。科学家对8 株链球菌(Streptococcus agalactiae)的基因组序列进行比较分析,结果显示泛基因组大小为2 713,其中1 806 个基因属核心基因(置信区间为95%),907 个基因属附属基因[3]。通过指数衰减模型拟合,可预测该物种每新增1 个基因组序列,将平均增加33 个新基因,表明链球菌拥有一个开放的泛基因组。同样,在30 株大肠埃希菌的研究中[7],也呈现一个开放的泛基因组,大小为2 786,其中核心基因为2 344 个(约占49.7%),菌株特异性基因为539个(约占19.3%)。从大肠埃希菌泛基因组拟合曲线可推断,每个新大肠埃希菌基因组完成测序平均约146 个新基因被发现。相反,在炭疽芽胞杆菌(Bacillus anthracis)的系统分析表明了一种不同的趋势[8],线性拟合结果显示,菌株特异性基因很快收敛至0,表明其拥有一个闭合的泛基因组。一般而言,泛基因组分类可反映物种基因组多样性的程度,体现该物种适应环境的能力及进化的快慢。
2 泛基因组研究策略
2.1 基因组的测序及组装 基因组测序技术是现代分子生物学研究中重要的技术,通过测序可精确且全面地反映基因组与转录组的遗传信息,科学地揭示物种的多样性及复杂性[9]。第二代测序技术在第一代测序技术基础上,通过分析方法的并行化、自动化及计算机化提高了测序通量,这种技术上的飞跃使批量获得基因组信息成为可能[10]。由于第二代测序技术存在一些局限性,表现在序列读长比较短,如 illumina 平台最长为 250 ~ 300 bp,454 平台最长约为500 bp;PCR 富集序列建库的过程中,含量较少的序列可能无法大量扩增,造成部分信息丢失。第三代测序技术一定程度上弥补了第二代的缺陷,以单分子为单位进行实时测序,不需进行PCR 扩增,进而可提供高质量组装参考的基因组序列[11]。泛基因组分析的科学性很大程度上取决于基因组组装及注释的准确性,足够完整的序列是一个先决条件。DNA测序技术及各种基因组组装、注释工具的不断发展推动泛基因组学分析进入了一个新的发展时期[12]。
微生物基因组学研究是实现片段化测序到完整基因组的组装,再到基因组遗传信息的解密。基因组测序及组装是泛基因组分析中的重要数据基础,序列组装的质量、完整性及精确性直接影响注释和分析的结果。组装旨在不引入错误的前提下创建最完整的重构。基因组组装的效果与测序的深度、覆盖度及组装的方法有关。测序深度与基因组覆盖度间呈密切相关的非线性关系,测序深度带来的基因错误检测率或假阳性检测结果随基因组测序深度的提升而下降。常用的测序组装工具有SOAPdenovo、Trimity 及 Abyss 等[13]。
2.2 直系同源序列的鉴定及基因组的注释 目前,在物种基因组学的分析中,最重要的工作之一是通过判断一段基因序列在该物种基因组中存在及分布的宽度,即多少菌株中有可能存在这一段基因序列。直系同源的基因(ortholog)即有共同来源的基因是物种形成过程中被复制保留下来具有相同功能的基因。直系同源基因通常被广泛认为是具有相似基因结构及生物学功能的基因,在绝大多数生物中具有负责编码核心基因的功能[14]。在泛基因组研究中,通过评估同源簇在菌株中的分布广度来定义核心基因组及非必需基因基因。直系同源基因的识别在基因功能注释、物种系统发育评估、系统发育树构建等分析中有广泛的应用[15]。
直系同源基因的识别主要通过生物信息学方法鉴定,进化关系越近的基因其结构及功能越相似[16]。在多菌株基因组分析中,主要采用序列比对的方法来判断序列间的同源关系。OrthoMCL 及OrthoFinder 是识别同源基因应用较多的两种工具[17-18]。OrthoMCL 利用马尔可夫聚类算法改善了多基因组比对固有的多对多的直系同源关系,允许在相似性空间中同时对全局关系进行分类,可快速且准确地处理复杂的结构域。OrthoFinder 分析中有两个选项:一个是序列搜索比对的方式,默认比对方法为DIAMOND[19],另一个是同源关系树的推断方法,默认选项为DendroBLAST[20]。该设置为用户选择分析方法提供了灵活性,针对不同分析对象可选择更适合的算法。OrthoFinder 是一款基于正交群推理算法的识别工具,其优点是高效、准确且可扩展到数千个基因组。输入文件为物种编码蛋白的氨基酸序列文件(FASTA 文件),整个分析仅需一行命令即可启动,操作方式简洁。
随着基因组分析技术的发展,基因组的结构动态及功能也倍受关注。基因组注释是确定基因在整个编码区域的位置及其生物学功能,主要步骤为:①基因组中功能区域序列的识别;②定位基因组中的元素(该过程称为基因预测);③将生物学信息附加至这些元素上。大多数注释工具需先在特定数据库进行同源基因搜索,再将同源基因信息对应至基因组上,如 RAST[21]、xBASE2[22]及 Prokka[23]。
2.3 泛基因组的模型构建 TETTELIN 等[2,6]率先提出了评估泛基因组大小、核心基因组及新基因发现率的算法。即向一个小的泛基因组模型(仅含2个基因组)里加基因组,会发现大量的新基因,因开始的基因库很小;相反,核心基因组的大小将减少,因基因被所有基因组共享的可能减小。被研究的基因组数目越多,泛基因组越大,核心基因组的大小减小,收敛至一个临界值,同时,新发现的基因数目将渐近地稳定在某个值上。对于一个闭合的泛基因组,新基因数目收敛于0,可估计其泛基因组的大小;对于一个开放的泛基因组,该值不为0,且无法估计泛基因组的大小(其可能无限增长)。在泛基因组分析中,核心基因的数量及菌株特异性基因的数量取决于有多少株菌株被分析,采用迭代方法,通过模拟所有可能组合(G 个基因组中),可推断核心基因组及菌株特异性基因的大小。对于n 个基因组(1 <n <G)的独立测量总数N 见下式。
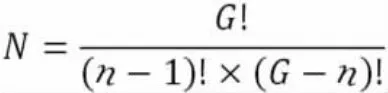
根据每个可能独立测量的结果,评估核心基因、菌株特异性基因的数量及泛基因组的大小。对一定数量的菌株进行分析时,核心基因组的大小及菌株特异性基因的数量被拟合外推指数衰减函数见下式。

式中Fc 及Fs 分别表示保守基因和菌株特异性基因的数目;n 为测序菌株的数量;κc、κs、τc、τs 及Ω 为自由参数;tg(θ)为代表泛基因组外推增长率的参数。泛基因组大小P(n)关于n 的函数见下式。

式中D 为输入基因组的平均基因数;P(n)拟合模型见下式。

通过将泛基因组大小、核心基因及特异性基因数目(以n 为函数)与上述指数函数进行拟合,得到最适拟合参数及其相关系数。测序新的基因组,发现新基因的数量将渐近达 tg(θ),因此,可将 tg(θ)值作为泛基因分类(开放或闭合)的依据。TETTELIN等[2]分析了8 个致病性链球菌的基因组并计算n株菌株所有可能的基因组集合,通过构建指数衰减模型推测8 株链球菌的核心基因的渐近值达1 806,新基因约为33 个。
2.4 常用分析包及工具 近几年,随着泛基因组分析技术的普及,大量分析工具被开发应用,它们在功能上有一定的相似性,能聚类同源基因、构建系统发育树及系统分析核心/ 非必需基因。这些工具在有各自特点的同时也存在一定的局限性,需不断地改进及完善[24]。
PGAP(Pan-Genomes Analysis Pipeline)是一款基于功能基因聚类的自动化集成分析软件[24],包括5 个功能模块,分别呈现全基因组图谱、物种进化、遗传变异、给定全基因组的不同菌株和分离株的功能富集,分析通过命令执行,集成性的过程极大提高了分析效率,其局限性在于输出文件缺乏直观性的文本文件。2014 年开发出带有图形界面的PanGP软件包倍受研究者的青睐[25]。PanGP 是为分析大规模细菌基因集合而开发的,除完全随机(totally random,TR)及距离指导(distance guide,DG)算法外,还提供了一种全面遍历算法(TA algorithm)。其输出结果以拟合的曲线图像呈现,通过非线性函数拟合了泛基因组、核心基因组及新基因集的数学模型。
此外,PAGE 等[26]开发了 Roary,一个典型的大规模构建原核生物泛基因组的工具。Roary 从基因组注释文件中提取编码区,并将其转换为蛋白质序列。一方面,过滤去除部分不完整序列,用CD-HIT进行迭代预聚类[27];另一方面,使用 BLASTP 对基因组序列进行相似性(默认参数为95%)比对[28],借助 MCL 对序列进行聚类[29];最后将 CD-HIT 中的预聚类结果与MCL 的结果合并一起,利用保守的基因邻域信息确定真正的同源簇。基于输入序列出现的顺序及在菌株中的覆盖率,对聚类的同源簇进行排序,根据同源簇对菌株进行聚类,菌株亲缘关系由聚类大小进行加权。该集成的程序使构建成千上万的原核生物基因组分析成为可能,并根据保守基因邻域信息进行更深的分析,保证了结果的准确性。
3 泛基因组分析的应用
泛基因组分析运用高通量测序及生物信息分析手段,构建泛基因组图谱,丰富该物种的遗传信息。目前研究趋势逐渐转向探索更大分类阶元的进化关系,作为比较基因组学的重要研究手段,泛基因组学从群体角度对细菌进行研究分析,根据细菌基因组动力学特征,剖析进化过程的动态变化[30]。此外,泛基因组分析通过比较不同菌株基因组及基因组成的差异,为细菌的耐药性、致病性及疫苗的制备等方面提供了重要的理论依据。目前,泛基因组已广泛用于深入了解肺炎链球菌[31]、流感嗜血杆菌[32]、大肠埃希菌[7,33]及枯草芽孢杆菌[34]等物种的研究。
泛基因组分析通过挖掘全基因集的遗传信息,深入全面了解特定物种的组成特征。为探索环状芽孢杆菌的次级代谢通路,姚彩苗等[35]借助泛基因组分析方法对9 个环状芽孢杆菌进行系统分析,发现泛基因组大小为9 572 个,核心基因组由3 622 个基因组成,特异性基因有4 593 个。通过对次级代谢产物合成基因簇分析,9 个环状芽孢杆菌基因组中共发现6 类、32 个次级代谢基因簇,重复出现最多的代谢通路是羊毛硫肽、套索肽及萜烯类化合物合成通路。该结果有助于更全面地了解环状芽孢杆菌,为其进一步研究提供了线索。
细菌种类通常根据基因含量、表型特征、生态位的性质及16S 核糖体核糖核酸序列来定义[36]。物种进化的主要分子机制是基因突变及基因重组现象。近年来,泛基因组分析也广泛应用于物种分类学中[37]。泛基因组重点分析基因组结构存在 / 缺失(presence / absence)的差异。泛基因组分析可通过两方面构建菌株间的进化关系:一方面选择单拷贝的核心基因,将它们按顺序联接进行多序列比对,构建系统发育关系;另一方面基于基因得失事件,不同菌株间基因比例及差异作为分析数据进行演化分析。泛基因组分析与传统的分类鉴定的方法相结合,可更准确地构建物种间的进化关系。
D′AURIA 等[38]对 5 个嗜肺军团菌(Legionella pneumophila)进行了系统的分析。嗜肺军团菌是一种细胞内的细菌,为一种人类病原体,该物种有一个开放的泛基因组。基于BLAST 的序列比对识别同源序列,确定核心基因组为1 979 个(占66.9%),非必需基因基因组大小为978 个(占33.1%)。核心基因组中大部分基因属基因组岛,共分为6 类:3 个与耐药相关的岛,1 个与重金属的分泌和运输相关的岛,3 个与 DNA 转移相关的岛,2 个 CRISPRs(clustered regularly interspaced short palindromic repeats)系统,7 个与噬菌体相关的系统,13 个功能不明的岛。结果表明,嗜肺军团菌的毒性及耐药性均为核心基因编码,这有利于进一步确定功能靶位点,为医学研发抗性药物提供理论基础。
4 小 结
细菌的基因组因其个体仅有几兆大小,先进的测序技术导致基因组数据大量的积累。泛基因组分析能更准确地反映细菌种类的概念,有助于全面揭示基因组的多样性。另外,通过对感染性细菌进行泛基因组分析,可挖掘毒力因子相关的基因,有助于医学研发相关制剂。预计在未来的几年里,收集到的细菌数据将超过实际能处理的数量,因此,需将泛基因组分析方法扩展到不同领域。本文为泛基因组分析的实际应用提供了参考。
